一种有机发光材料的合成工艺的制作方法
1.本发明涉及化工领域,特别是涉及一种有机发光材料1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺。
背景技术:
[0002]
随着有机发光材料的快速发展,尤其是有机发光材料基础的应用研究,通过化学合成的单体材料得到了广泛的运用。因此,一种简单、经济、安全、高效的合成1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑 的方法具有广泛的研究价值。
[0003]
目前,关于1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的制备方法的文献报道较少,方法也是千篇一律,主要以1-(4-溴苯基)-2-苯基-1h-1,3-苯二唑为原料,先用正丁基锂拔溴,与硼酸三甲酯反应生成化合物 2,再和smb做铃木偶联反应得到目标化合物的方法为主。该方法的缺点在于,合成步骤中需要用到危险、易燃的正丁基锂作为反应试剂;反应条件需要降温到-78℃,条件苛刻,化合物2的溶解性差,纯化困难。因而现有的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的制备方法,操作难度大,因而需要对其进行改进。
技术实现要素:
[0004]
本发明的目的在于针对背景技术中所述的现有技术中,1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,需要采用危险、易燃的正丁基锂,反应温度很低,需要降温到-78℃,中间产物溶解性差,纯化困难等问题,提供一种1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺。
[0005]
为实现以上目的,本发明通过以下技术方案予以实现:一种有机发光材料的合成工艺,其包括以下步骤:s1、在保护气氛下,向反应釜中加入溶剂,边搅拌边加入1-(4-溴苯基)-2-苯基-1h-1,3-苯二唑、联硼酸频哪醇酯和乙酸钾,加入催化剂,进行回流反应,监测1-(4-溴苯基)-2-苯基-1h-1,3-苯二唑反应完后降温,然后进行后处理,纯化得到2-苯基-1-[4-(四甲基-1,3,2-二氧苯甲醛-2-基)苯基]-1h-1,3-苯二唑;s2、在保护气氛下,向空反应釜中加入溶剂,边搅拌边加入步骤s1的产物2-苯基-1-[4-(四甲基-1,3,2-二氧苯甲醛-2-基)苯基]-1h-1,3-苯二唑、9-溴-10-(萘-2-基)蒽、碱和水,进行回流反应,监测2-苯基-1-[4-(四甲基-1,3,2-二氧苯甲醛-2-基)苯基]-1h-1,3-苯二唑和9-溴-10-(萘-2-基)蒽反应完成后降温,然后进行后处理,得到有机发光材料1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑。
[0006]
在上述方案的步骤s1中,后处理的方法为,降温至预定温度后,向反应液中加入水,然后用乙酸乙酯萃取,有机相旋蒸得到固体粗品,固体粗品用乙酸乙酯溶解,然后向其中滴加石油醚,搅拌1.5-3小时,降温至10-15℃,抽滤得到固体并烘干。
[0007]
在上述方案的步骤s2中,降温至预定温度后,对反应液抽滤得到固体粗品,固体粗
品用四氢呋喃溶解,加入活性炭,搅拌1.5-3小时,过滤得到液体,旋除溶剂,得到固体产物并烘干。过滤时,可以采用硅胶柱过滤,也可以采用抽滤漏斗垫上硅胶进行过滤,得到黄色液体,之后将母液浓缩干,得到固体产物。
[0008]
在上述方案的步骤s1中,所述溶剂为1,4-二氧六环。
[0009]
在上述方案的步骤s1中,所述1-(4-溴苯基)-2-苯基-1h-1,3-苯二唑、联硼酸频哪醇酯和乙酸钾的当量比为:1.0:(1.0-1.5):(2.0-3.0)。
[0010]
在上述方案的步骤s1中,加热至回流反应的温度为100-120℃。
[0011]
在上述方案的步骤s2中,所述溶剂为甲苯。
[0012]
在上述方案的步骤s2中,1-(4-溴苯基)-2-苯基-1h-1,3-苯二唑、9-溴-10-(萘-2-基)蒽、碱和水的当量比为:1.0:(1.0-1.5):(2.0-3.0):(2.0-4.0)。
[0013]
在上述方案的步骤s2中,所述的碱为碳酸钾、碳酸钠、磷酸钾、磷酸钠或其他碱性金属盐。
[0014]
在上述方案的步骤1和步骤2反应完成后,降温至30℃以下,进行后处理。
[0015]
在上述方案的步骤s1中所述的催化剂为钯催化剂。
[0016]
本发明具有积极的效果:1)本发明的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,合成原料中,避免了采用危险、易燃的正丁基锂,而是采用了联硼酸频哪醇酯,使合成工艺的安全性很高;2)本发明的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,采用了加热回流工艺,避免了采用-78℃的低温反应条件,合成工艺难度降低,更容易实施;3)本发明的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,制成的中间产物,溶解性好,通过简单的后处理即可完成纯化,能提高制成的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的纯度。
附图说明
[0017]
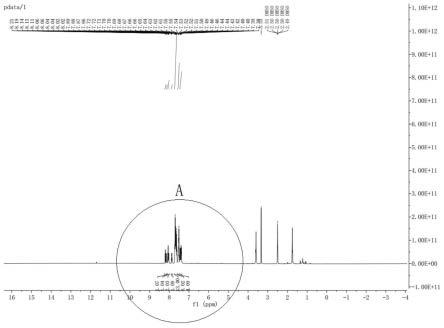
图1为本发明的实施例1的终产品的核磁检测结果。
[0018]
图2为图1中的a部的局部放大图。
[0019]
图3为本发明的实施例1的终产品的hplc检测结果。
[0020]
图4为本发明的实施例2的终产品的hplc检测结果。
[0021]
图5为本发明的实施例3的终产品的hplc检测结果。
[0022]
图6为本发明的实施例4的终产品的hplc检测结果。
具体实施方式
[0023]
下面通过实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0024]
实施例11-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,其包括以下步骤:
s1、向500 ml反应釜中加入1,4-二氧六环70 ml,搅拌下加入1-(4-溴苯基)-2-苯基-1h-1(下文中简称为sma),3-苯二唑 10g,联硼酸频哪醇酯7.64 g,乙酸钾56.20g,1,4-二氧六环10 ml洗涮残留物料,氮气置换3次,加入pd (dppf)cl2催化剂 220 mg,氮气置换3次,110℃加热回流4小时,hplc监测sma反应完全,降温至30℃以下。
[0025]
、反应液加200 ml水,用乙酸乙酯萃取,有机相旋蒸得到固体粗品,粗品在50℃用5倍体积乙酸乙酯溶解,滴加入50 倍体积的石油醚,50℃搅拌2小时,降温至10-15℃,抽滤得2-苯基-1-[4-(四甲基-1,3,2-二氧苯甲醛-2-基)苯基]-1h-1,3-苯二唑(下文中简称为p1)的固体烘干10.2 g,hplc纯度99.0%,收率96.9%。
[0026]
、氮气通入情况下,向500 ml的空反应釜中加入甲苯70 ml,搅拌下加入步骤s2中获得的p1 11 g,加入9-溴-10-(萘-2-基)蒽(下文简称为smb) 9.6 g, 加入乙醇80 ml,碳酸钾7.90 g,水 40 ml,加热至回流,反应12h,hplc监测p1少于5%,降温至30℃。
[0027]
、对反应液抽滤得到固体粗品,粗品用10倍体积的四氢呋喃溶解,加入粗品质量5%的活性炭,50℃搅拌2小时,滤硅胶柱,得到黄色液体,旋除溶剂,固体烘干,得到1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑14.65 g,hplc检测,其纯度为99.9%,通过计算可以得到产品收率为92%。
[0028]
本发明的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,其具体的反应方程为:通过上述的反应方程,可以看出本发明的合成工艺的工艺路线。本发明的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,合成原料中,避免了采用危险、易燃的正丁基锂,而是采用了联硼酸频哪醇酯,联硼酸频哪醇酯没有易燃易爆等危险,因而本发明的合成工艺的安全性很高;本发明的合成工艺,反应温度控制在110℃,避免了采用-78℃的低温反应条件,合成工艺难度降低,更容易实施;本发明的合成工艺,制成的中间产物,溶解性好,通过简单的后处理即可完成纯化,能提高制成的1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的纯度。
[0029]
如图1和2所示,为本发明的实施例1的终产品的核磁检测结果和hplc检测结果,通过图1和图2可以看出,核磁干净,无明显杂峰,通过图3可以得出,hplc检测,产品纯度为99.9%。
[0030]
实施例21-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,其包
括以下步骤:s1、向5 l反应釜中加入1,4-二氧六环700 ml,搅拌下加入sma 100g,联硼酸频哪醇酯76.4 g,乙酸钾562.0g,1,4-二氧六环100 ml洗涮残留物料,在氮气氛围下,加入pd (dppf)cl2催化剂 2.2 g,在氮气氛围下,110℃加热回流4小时,hplc监测sma反应完全,降温至30 oc
以下。
[0031]
、反应液中加入200 ml水,用乙酸乙酯萃取,有机相旋蒸得到固体粗品,粗品在50℃用5倍体积乙酸乙酯溶解,滴加入50 倍体积的石油醚,50℃搅拌2小时,降温至10-15℃,抽滤烘干得10.2 g的固体产物p1,hplc检测,其纯度为99.0%,通过计算可以得到产品收率为96.9%。
[0032]
、氮气通入情况下,向5 l反应釜中加入甲苯700 ml,搅拌下加入110.0 g 的p1,加入96.0 g 的smb, 加入乙醇800 ml,碳酸钾79.0 g,水 400 ml,加热至回流,反应15小时,hplc监测p1少于 5%,降温至30℃以下。
[0033]
、对反应液抽滤得到固体粗品,粗品用10倍体积四氢呋喃溶解,加入粗品质量5%的活性炭,50℃搅拌2小时,用硅胶柱过滤,得到黄色液体,旋除溶剂,固体烘干,得到1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑148 g,如图4所示,hplc检测,其纯度为99.9%,收率为92.1%。
[0034]
实施例31-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,其包括以下步骤:s1、向50 l反应釜中加入1,4-二氧六环7 l,搅拌下加入sma 1kg,联硼酸频哪醇酯764 g,乙酸钾5620g,1,4-二氧六环1 l洗涮残留物料,在氮气氛围加入pd (dppf)cl2催化剂22 g,氮气氛围下,110℃加热回流4.5小时,hplc监测sma反应完全,降温至30 oc
以下。
[0035]
、向反应液中加入20l水,用乙酸乙酯萃取,有机相旋蒸得到固体粗品,粗品在50℃用5倍体积乙酸乙酯溶解,滴加入50 倍体积的石油醚,50℃搅拌2小时,降温至10-15℃,抽滤烘干得1053 g的固体产物p1,hplc检测,其纯度为99.0%,通过计算可以得到产品收率为97.0%。
[0036]
、在氮气通入情况下,向500 ml反应釜中加入甲苯70 ml,搅拌下加入1100 g的p1,加入960 g 的smb, 加入乙醇8 l,碳酸钾790 g,水 4 l, 加热至回流,反应16小时,hplc监测p1少于 5%,降温至30℃以下。
[0037]
、对反应液抽滤得到固体粗品,粗品用10倍体积四氢呋喃溶解,加入粗品质量5%的活性炭,50℃搅拌2小时,用硅胶柱过滤,得到黄色液体,旋除溶剂得固体烘干旋除溶剂,固体烘干,得到1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑1482 g,如图5所示,hplc检测,其纯度为99.9%,通过计算可以得到产品收率为92.1%。
[0038]
实施例41-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑的合成工艺,其包括以下步骤:s1、向100 l反应釜中加入1,4-二氧六环35l,搅拌下加入sma 5 kg,联硼酸频哪醇酯3820 g,乙酸钾28.1kg,1,4-二氧六环5 l洗涮残留物料,氮气置换3次,加入pd (dppf)cl2催化剂1.1 kg,氮气置换3次,110℃加热回流4小时,hplc监测sma反应完全,降温至30 oc
以下。
[0039]
、向反应液中加入20l水,用乙酸乙酯萃取,有机相旋蒸得到固体粗品,粗品在50℃用5倍体积乙酸乙酯溶解,滴加入50 倍体积的石油醚,50℃搅拌2小时,降温至10-15℃,抽滤烘干得5300 g的固体产物p1,hplc检测,其纯度为99.0%,收率为96.9%。
[0040]
、氮气通入情况下,向100 l反应釜中加入甲苯35l,搅拌下加入5.5 kg的p1,加入4.8 kg的smb, 加入乙醇10l,碳酸钾7.90 g,水 10l,加热至回流,反应20小时,hplc监测p1少于 5%,降温至30℃以下。
[0041]
、对反应液抽滤得到固体粗品,粗品用10倍体积四氢呋喃溶解,加入粗品质量5%的活性炭,50℃搅拌2小时,用硅胶柱过滤,得到黄色液体,旋除溶剂,固体烘干,得到1-(4-(10-(萘-2-基)蒽-9-基)苯基)-2-苯基-1h-苯并[d]咪唑7420 g,如图6所示,hplc检测,其纯度为99.9%,通过计算可以得到产品收率为92.6%。
[0042]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1