一种β-(2-吲哚基)丙醛衍生物的合成方法
一种
β-(2-吲哚基)丙醛衍生物的合成方法
技术领域
1.本发明涉及有机合成技术领域,具体涉及一种β-(2-吲哚基)丙醛衍生物的合成方法。
背景技术:
2.吲哚是各种生物活性化合物中最具药用价值的杂环化合物之一。吲哚基团是许多天然产物以及药物分子的重要骨架,发展吲哚类化合物的高效制备反应是有机合成领域的研究热点之一。目前,已经报道的关于吲哚c2的羰乙基化反应的研究相对较少,而如何经济、快速、高效地构建功能性醛基化反应备受关注。
3.近年来,实现c-c键的构建主要包括以下两种策略:
4.1)在过渡金属催化下,通过利用不同导向基团实现芳基csp
2-h羰乙基化反应,例如:a)以α,β不饱和醛/酮及其衍生物为偶联试剂(yoshino,t.;ikemoto,h.;matsunaga,s.;kanai,m.acationic high-valent cp*coiii complex for the catalytic generation of nucleophilic organometallic species:directed c-h bond activation.[j]angew.chem.,int.ed.2013,125,2263;li,j.;zhang,z.;ma,w.;tang,m.;wang,d.;zou,l.-h.mild cobalt(iii)-catalyzed c-hhydroarylation of conjugated c=c/c=o bonds.[j]adv.synth.catal.2017,359,1717);b)以廉价金属锰为催化剂,重氮为偶联试剂(lu,q.;mondal,s.;cembellin,s.;glorius,f.mn(i)/ag(i)relay catalysis:traceless diazo-assisted csp
2-h/csp
3-h coupling toβ-(hetero)aryl/alkenyl.[j]angew.chem.,int.ed.2018,57,10732);
[0005]
2)利用c-c键活化的策略,先通过金属对目标c-c键进行切割,然后再原位实现羰乙基化反应(liu,z.;hu,x.;yang,c.;xie,h.;jiang,h.;zeng,w.rh(iii)-catalyzed csp2-csp3bond cleavage/carbonylethylation of alpha-indolyl alcohols.[j]adv.synth.catal.2021,363,1672)。
[0006]
然而,上述方法实现的均是分子间的羰乙基化反应,能够有效地合成目标产物,但需要使用的偶联试剂的当量普遍较高,原子经济性低,且大部分反应用到的多为贵金属,同时反应条件较为苛刻,应用受到很大限制。
[0007]
β-(2-吲哚基)丙醛作为潜在药物分子骨架和天然产物骨架分子具有很好的应用前景,因此开发一种具有原料廉价、简便高效、反应条件温和、原子经济性高、产物易纯化、绿色经济等优点的β-(2-吲哚基)丙醛衍生物合成方法具有十分重要的意义。
技术实现要素:
[0008]
本发明的目的在于提供一种β-(2-吲哚基)丙醛衍生物的合成方法。
[0009]
本发明所采取的技术方案是:
[0010]
一种β-(2-吲哚基)丙醛衍生物的合成方法包括以下步骤:将n-吡啶吲哚烯丙醇类化合物和催化剂分散在有机溶剂中进行分子重排反应,即得β-(2-吲哚基)丙醛衍生物;所
述n-吡啶吲哚烯丙醇类化合物的结构式如式a所示;所述β-(2-吲哚基)丙醛衍生物的结构如式b所示:
[0011][0012]
式a和式b中,r1选自-h、苯基、4-甲基苯基、4-氯苯基中的一种,r2选自甲基、-cl、-br、-cf3中的一种,r3选自-h、甲基、-f、-br、甲氧基中的一种。
[0013]
优选的,所述n-吡啶吲哚烯丙醇类化合物、催化剂的摩尔比为1:0.04~0.06。
[0014]
优选的,所述催化剂为五羰基溴化锰(i)、二氯(五甲基环戊二烯基)合铑(iii)二聚体、二(六氟锑酸)三乙腈(五甲基环戊二烯基)铑(iii)中的一种。
[0015]
进一步优选的,所述催化剂为五羰基溴化锰(i)。
[0016]
优选的,所述丙醇类化合物、有机溶剂的摩尔比为1:100~200。
[0017]
优选的,所述有机溶剂为1,2-二氯乙烷、甲苯、三氟甲苯中的至少一种。
[0018]
优选的,所述分子重排反应在45℃~75℃下进行。
[0019]
优选的,所述分子重排反应的时间为12h~48h。
[0020]
优选的,所述β-(2-吲哚基)丙醛衍生物通过柱层析进行分离纯化。
[0021]
优选的,所述柱层析采用的洗脱液由石油醚和乙酸乙酯组成。
[0022]
本发明的有益效果是:本发明的β-(2-吲哚基)丙醛衍生物合成方法具有原料廉价、简便高效、反应条件温和、原子经济性高、产物易纯化、绿色经济等优点,且该合成方法对空气和水都不敏感,得到的产物可以作为潜在药物直接应用到医药领域中,应用前景广阔。
附图说明
[0023]
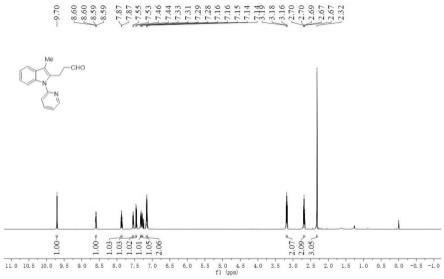
图1为实施例1的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0024]
图2为实施例1的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0025]
图3为实施例2的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0026]
图4为实施例2的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0027]
图5为实施例3的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0028]
图6为实施例3的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0029]
图7为实施例4的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0030]
图8为实施例4的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0031]
图9为实施例5的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0032]
图10为实施例5的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0033]
图11为实施例6的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0034]
图12为实施例6的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
[0035]
图13为实施例7的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图。
[0036]
图14为实施例7的β-(2-吲哚基)丙醛衍生物的核磁共振碳谱图。
具体实施方式
[0037]
下面结合具体实施例对本发明作进一步的解释和说明。
[0038]
实施例1~7中的n-吡啶吲哚烯丙醇类化合物参照以下方法制备而成(参考文献:s.;mclean,e.;donnelly,l.;hockin,b.;taylor,j.,arylboronic acid catalyzed c-alkylation and allylation reactions using benzylic alcohols.[j]org.lett.2020,22,7547-7551):将1.0mmol的n-吡啶吲哚醛s(参考文献:zhu,c.;pinkert,t.;greβies,s.;glorius,f.,one-pot c-h formylation enabled by relay catalysis of manganese(i)and iron(iii).[j]acs catal.2018,8,10036-10042)和10ml的无水四氢呋喃(thf)加入三颈圆底烧瓶中,在氩气气氛中溶解,再将溶液冷却至0℃后滴加2.0ml浓度为1.0mol/l的乙烯基溴化镁的thf溶液,加完后升温至室温并搅拌过夜,再用饱和nh4cl溶液淬灭反应,再用乙酸乙酯萃取反应液3次(每次使用10ml的乙酸乙酯),合并有机层,用无水na2so4干燥,再抽真空蒸发溶剂得到粗产物,再在硅胶柱上用石油醚/乙酸乙酯(体积比10:1~5:1)快速分离纯化,得到n-吡啶吲哚烯丙醇类化合物,反应式如下:
[0039][0040]
实施例1:
[0041]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0042]
将52.8mg(0.2mmol)的1-(2-吡啶基)-2-(1-羟基烯丙基)-3-甲基吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到42.0mg的β-(2-吲哚基)丙醛衍生物(产率:80%)。
[0043]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图1所示,核磁共振碳谱图如图2所示。
[0044]
谱图分析:
[0045]1h nmr(500mhz,cdcl3):δ9.70(s,1h),8.60(dd,j=4.7,1.4hz,1h),7.87(td,j=7.7,1.9hz,1h),7.54(dd,j=5.5,3.4hz,1h),7.45(d,j=8.0hz,1h),7.32(dd,j=5.8,3.3hz,1h),7.28(dd,j=7.2,5.1hz,1h),7.17-7.13(m,2h),3.21-3.16(m,2h),2.71-2.66(m,2h),2.32(s,3h)。
[0046]
13
c nmr(126mhz,cdcl3):δ201.51,151.55,149.63,138.39,136.41,134.98,129.41,122.20,121.83,120.68,120.45,118.44,110.53,109.89,43.89,17.96,8.75。
[0047]
hr-ms:理论值[m+h]
+
:c
17h17
n2o:265.1335,测量值:265.1339。
[0048]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0049][0050]
实施例2:
[0051]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0052]
将65.2mg(0.2mmol)的1-(2-吡啶基)-2-(1-羟基烯丙基)-3-苯基吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到46.3mg的β-(2-吲哚基)丙醛衍生物(产率:71%)。
[0053]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图3所示,核磁共振碳谱图如图4所示。
[0054]
谱图分析:
[0055]1h nmr(500mhz,cdcl3):δ9.52(s,1h),8.65(dd,j=4.8,1.3hz,1h),7.93(td,j=7.8,1.9hz,1h),7.59(d,j=7.3hz,1h),7.54(d,j=8.0hz,1h),7.49(dt,j=15.2,7.3hz,4h),7.35(td,j=7.1,2.6hz,3h),7.21-7.14(m,2h),3.32-3.26(m,2h),2.56(dd,j=11.2,4.1hz,2h)。
[0056]
13
c nmr(126mhz,cdcl3):δ201.05,151.29,149.85,138.62,136.65,135.36,134.64,129.94,128.73,128.29,126.74,122.70,122.43,121.21,121.17,119.31,117.97,110.08,43.93,18.21。
[0057]
hr-ms:理论值[m+h]
+
:c
22h19
n2o:327.1492,测量值:327.1488。
[0058]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0059][0060]
实施例3:
[0061]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0062]
将59.6mg(0.2mmol)的1-[2-(5-氯吡啶基)]-2-(1-羟基烯丙基)-3-甲基吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到41.7mg的β-(2-吲哚基)丙醛衍生物(产率:70%)。
[0063]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图5所示,核磁共振碳谱图如图6所示。
[0064]
谱图分析:
[0065]1h nmr(400mhz,cdcl3):δ9.76(s,1h),8.57(d,j=2.6hz,1h),7.88(dd,j=8.5,2.6hz,1h),7.59-7.56(m,1h),7.46(d,j=8.5hz,1h),7.33(dt,j=6.4,2.8hz,1h),7.22-7.18(m,2h),3.21-3.16(m,2h),2.75(dd,j=11.1,4.1hz,2h),2.34(s,3h)。
[0066]
13
c nmr(101mhz,cdcl3):δ201.37,149.70,148.35,138.21,136.21,134.90,129.72,129.51,122.46,121.15,120.77,118.59,111.07,109.73,43.92,17.88,8.76。
[0067]
hr-ms:理论值[m+h]
+
:c
17h16
cln2o:299.0946,测量值:299.0944。
[0068]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0069][0070]
实施例4:
[0071]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0072]
将68.4mg(0.2mmol)的1-[2-(5-溴吡啶基)]-2-(1-羟基烯丙基)-3-甲基吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到45.0mg的β-(2-吲哚基)丙醛衍生物(产率:66%)。
[0073]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图7所示,核磁共振碳谱图如图8所示。
[0074]
谱图分析:
[0075]1h nmr(500mhz,cdcl3):δ9.72(s,1h),8.63(d,j=2.2hz,1h),7.97-7.95(m,1h),7.55-7.52(m,1h),7.36(d,j=8.5hz,1h),7.32-7.29(m,1h),7.18-7.15(m,2h),3.15(t,j=7.6hz,2h),2.71(t,j=7.6hz,2h),2.30(s,3h)。
[0076]
13
c nmr(126mhz,cdcl3):δ201.3,150.6,150.1,141.0,136.2,134.9,129.6,122.5,121.6,120.8,118.6,118.0,111.2,109.8,43.9,17.9,8.8。
[0077]
hr-ms:理论值[m+h]
+
:c
17h16
brn2o:343.0441,测量值:343.0438。
[0078]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0079][0080]
实施例5:
[0081]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0082]
将56.0mg(0.2mmol)的1-(2-吡啶基)-2-(1-羟基烯丙基)-5-甲氧基吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应
24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到39.8mg的β-(2-吲哚基)丙醛衍生物(产率:71%)。
[0083]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图9所示,核磁共振碳谱图如图10所示。
[0084]
谱图分析:
[0085]1h nmr(400mhz,cdcl3):δ9.79(s,1h),8.65(dd,j=4.9,1.2hz,1h),7.92-7.87(m,1h),7.46(d,j=8.0hz,1h),7.34-7.31(m,1h),7.27(d,j=8.9hz,1h),7.07(d,j=2.4hz,1h),6.82(dd,j=8.9,2.5hz,1h),6.39(s,1h),3.87(s,3h),3.20(t,j=7.5hz,2h),2.86-2.82(m,2h)。
[0086]
13
c nmr(101mhz,cdcl3):δ201.4,154.9,151.3,149.7,140.0,138.5,132.3,129.1,122.0,120.6,111.7,111.0,102.6,102.3,55.8,42.9,20.5。
[0087]
hr-ms:理论值[m+h]
+
:c
17h17
n2o2:281.1285,测量值:281.1276。
[0088]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0089][0090]
实施例6:
[0091]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0092]
将53.6mg(0.2mmol)的1-(2-吡啶基)-2-(1-羟基烯丙基)-5-氟吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到38.7mg的β-(2-吲哚基)丙醛衍生物(产率:72%)。
[0093]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图11所示,核磁共振碳谱图如图12所示。
[0094]
谱图分析:
[0095]1h nmr(400mhz,cdcl3):δ9.80(s,1h),8.68-8.66(m,1h),7.95-7.91(m,1h),7.46(d,j=8.0hz,1h),7.39-7.35(m,1h),7.29-7.21(m,2h),6.92-6.87(m,1h),6.41(s,1h),3.18(t,j=7.4hz,2h),2.88-2.84(m,2h)。
[0096]
13
c nmr(101mhz,cdcl3):δ201.1,158.5(d,j=236.3,1j
cf
),151.0,149.8,141.1,138.6,133.8,128.9(d,j=10.1,3j
cf
),122.5,120.9,110.9(d,j=9.1,3j
cf
),110.1(d,j=26.3,2j
cf
),105.2(d,j=23.2,2j
cf
),102.5(d,j=4.0,3j
cf
),42.8,20.3。
[0097]
hr-ms:理论值[m+h]
+
:c
16h14
fn2o:269.1085,测量值:269.1078。
[0098]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0099]
[0100]
实施例7:
[0101]
一种β-(2-吲哚基)丙醛衍生物的合成方法,其包括以下步骤:
[0102]
将65.6mg(0.2mmol)的1-(2-吡啶基)-2-(1-羟基烯丙基)-5-溴吲哚、2.74mg(0.01mmol)的五羰基溴化锰(i)和2ml的1,2-二氯乙烷(分析纯)加入封管中,75℃搅拌反应24h,冷却至室温,旋干,采用柱层析进行分离纯化,洗脱液由石油醚和乙酸乙酯按照体积比5:1组成,得到42.8mg的β-(2-吲哚基)丙醛衍生物(产率:65%)。
[0103]
本实施例的β-(2-吲哚基)丙醛衍生物的核磁共振氢谱图如图13所示,核磁共振碳谱图如图14所示。
[0104]
谱图分析:
[0105]1h nmr(400mhz,cdcl3):δ9.80(s,1h),8.67(dd,j=4.8,1.0hz,1h),7.95-7.91(m,1h),7.70(d,j=1.5hz,1h),7.44(d,j=7.6hz,1h),7.40-7.35(m,1h),7.25-7.18(m,2h),6.38(s,1h),3.17(t,j=7.4hz,2h),2.85(t,j=7.5hz,2h)。
[0106]
13
c nmr(101mhz,cdcl3):δ201.0,150.8,149.9,140.8,138.7,135.9,130.1,124.8,122.6,120.9,114.0,111.6,101.9,42.7,20.2。
[0107]
hr-ms:理论值[m+h]
+
:c
16h14
brn2o:329.0284,测量值:329.0279。
[0108]
综上可知,本实施例的β-(2-吲哚基)丙醛衍生物的结构式如下:
[0109][0110]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1