一种嘧啶酮类化合物及其制备方法和医药应用
1.本发明属于药物化学和药物治疗学技术领域,具体涉及一种嘧啶酮类化合物。该类化合物可用于制备与pak5有关疾病的药物。本发明还涉及该类化合物的制备方法以及含有它们的药物组合。
背景技术:
2.p-21激活激酶(paks)是小g蛋白家族成员cdc42和rac的重要下游效应器,与人类相关的paks家族成员按序列和结构同源性分为i类(pak1、pak2、pak3)和ii类(pak4、pak5、pak6)两类。i类paks和ii类paks具有50%的同源性,ii类paks(pak4、pak5、pak6)具有80%的同源性,均具有保守的c端的激酶域和n端的调节域,n端有1个自身磷酸化区域(p21 gtpase binding domain,pbd)。通过分析paks蛋白晶体结构发现:相对于i类pak激酶,ii型paks激酶n端不存在pid抑制区域,因此激酶活性更高。ii型paks的激酶活性主要由其c端的激酶催化域决定,而pak5被认为是肿瘤发展的重要介导物,从各亚型结构来看,pak5在肿瘤发展过程中显示出其特有的功能和机制。
3.作用机制研究发现p21激活激酶5(pak5)可定位于线粒体,磷酸化bad蛋白的ser112位点引发肿瘤细胞抗凋亡;磷酸化nf-κb的p65亚基,促进p65的核转位,进而上调细胞周期素cyclind1的表达,促进乳腺癌细胞在体内外的增殖;磷酸化gata-1抑制乳腺癌细胞上皮间质转化;磷酸化e47抑制结肠癌转移;磷酸化p120-catenin-ser288复合物引起细胞骨架重组,促进肿瘤细胞运动。
4.pak5小分子抑制剂对于治疗涉及pak5高表达的癌症有良好前景。目前,有关pak5小分子抑制剂的报道较少,其中包括辉瑞制药研发的pf-3758309以及基因泰克公司研发的gne-2861等。
技术实现要素:
5.本发明的目的是在现有技术的基础上,提供一种嘧啶酮类化合物,药理实验证明,该类化合物具有良好的pak5抑制活性,具体对肾癌细胞、肝癌细胞、结肠癌细胞和乳腺癌细胞等肿瘤细胞也有较强的抗肿瘤活性。
6.本发明的另一目的是提供一种上述化合物的制备方法。
7.本发明的另一目的是提供一种上述化合物在医药方面的用途。
8.本发明的技术方案如下:
9.本发明涉及结构如通式i所示的化合物、异构体、水合物、溶剂化物或其药学上可接受的盐,
[0010][0011]
其中,
[0012]
r1代表氢或c
1-c6烷基;
[0013]
r2代表氢、卤素、氰基、三氟甲基、羟基、氨基、硝基、c
1-c6烷基、c
1-c6烷氧基、苯基、取代苯基、苯氧基、取代苯氧基、苄基或取代苄基,所述的取代苯基、取代苯氧基或取代苄基可任意地由下述取代基单取代或多取代:氰基、三氟甲基、羟基、氨基、硝基、c
1-c4烷基或c
1-c4烷氧基;
[0014]
l代表共价键、-ch
2-、-ch
2 ch
2-、-nh-ch
2-、-ch
2-nh-、-nh-co-或-co-nh-;
[0015]
r3代表卤素、羟基、c
1-c4烷氧基、c
1-c4烷硫基、ch
3-so
2-或ch2ch
3-so
2-;
[0016]
n代表1-3的整数。
[0017]
在一种优选方案中,r1代表氢、甲基、乙基、异丙基或叔丁基。
[0018]
在一种更优选方案中,r1代表氢、甲基或乙基。
[0019]
在一种优选方案中,r2代表氢、三氟甲基、氟、氯、氰基、甲基、乙基、甲氧基、乙氧基、苯氧基或苄基。
[0020]
在一种更优选方案中,r2代表氢、三氟甲基或苯氧基。
[0021]
在一种优选方案中,r3代表氟、氯、羟基、甲氧基、乙氧基、甲硫基、乙硫基或ch
3-so
2-。
[0022]
在一种更优选方案中,r3代表氟、羟基、甲氧基、乙氧基、甲硫基或ch
3-so
2-。
[0023]
在一种优选方案中,l代表共价键、-ch
2-或-ch
2 ch
2-。
[0024]
在一种优选方案中,n代表1或2。
[0025]
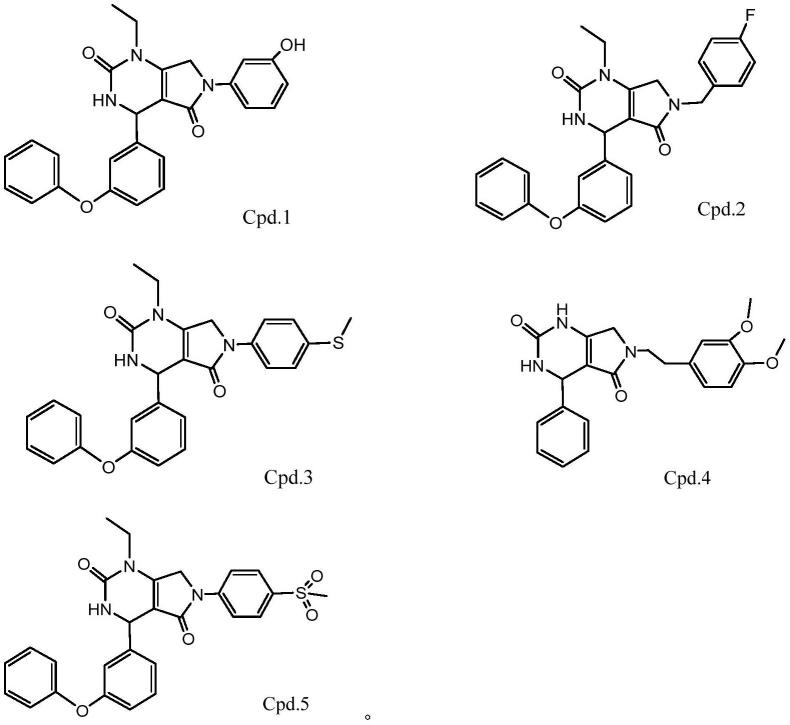
进一步地,通式i所述化合物优选自下列化合物:
[0026][0027]
本发明公开了一种通式i所述化合物的制备方法,其合成路线如下:
[0028]
[0029][0030]
优选地,反应条件如下:第一步中,反应条件为tsoh、etoh和回流;第二步中,反应条件为aibn、ccl4和回流;第三步中,反应条件为et3n、meoh和回流。
[0031]
这些中间体或目标化合物均可按照常规分离技术加以纯化,并且根据需要将其转化为与可药用酸的加成盐。
[0032]
本发明还提供了一种药物组合物,它以上述的化合物、异构体、水合物、溶剂化物或其药学上可接受的盐为活性成分或主要活性成分,辅以药学上可接受的辅料。其中,药物组合物可以为片剂、胶囊、颗粒剂、注射剂或喷雾剂。所述药学上可接受的载体选自填充剂、崩解剂、粘合剂和润滑剂中的一种或多种,包括但不限于任何和全部的溶剂、分散介质、包衣、吸收延迟剂等。
[0033]
本发明提供的嘧啶酮类化合物,具有良好的pak5抑制活性,可以作为pak5抑制剂,用于制备与pak5有关疾病的药物中,其中,pak5有关疾病为结直肠癌、肝癌、胃癌、宫颈癌、肾癌、乳腺癌或糖尿病。
[0034]
除非另外说明,在说明书和权利要求中使用的以下术语具有下面讨论的含义:
[0035]“药学上可接受的盐”表示保留母体化合物的生物有效性和性质的那些盐。这类盐包括:
[0036]
(1)与酸成盐,通过母体化合物的游离碱与无机酸或有机酸的反应而得,无机酸包括盐酸、氢溴酸、硝酸、磷酸、偏磷酸、硫酸、亚硫酸和高氯酸等,有机酸包括乙酸、三氟乙酸、丙酸、丙烯酸、己酸、环戊烷丙酸、羟乙酸、丙酮酸、草酸、(d)或(l)苹果酸、富马酸、马来酸、抗坏血酸、樟脑酸、苯甲酸、羟基苯甲酸、γ-羟基丁酸、甲氧基苯甲酸、邻苯二甲酸、甲磺酸、乙磺酸、萘-1-磺酸、萘-2-磺酸、对甲苯磺酸、水杨酸、酒石酸、柠檬酸、乳酸、肉桂酸、十二烷基硫酸、葡糖酸、谷氨酸、天冬氨酸、硬脂酸、扁桃酸、琥珀酸、戊二酸或丙二酸等。
[0037]
(2)存在于母体化合物中的酸性质子被金属离子代替或者与有机碱配位化合所生成的盐,金属例子例如碱金属离子、碱土金属离子或铝离子,有机碱例如乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n-甲基葡糖胺、奎宁等。
[0038]“药物组合物”指将本发明中的化合物中的一个或多个或其药学上可接受的盐、溶剂化物、水合物或前药与别的化学成分,例如药学上可接受的载体,混合。药物组合物的目的是促进给药给动物的过程。
[0039]“药用载体”或“药学上可接受的载体”指的是对有机体不引起明显的刺激性和不干扰所给予化合物的生物活性和性质的药物组合物中的非活性成分,例如但不限于:碳酸钙、磷酸钙、各种糖(例如乳糖、甘露醇等)、淀粉、环糊精、硬脂酸镁、纤维素、碳酸镁、丙烯酸聚合物或甲基丙烯酸聚合物、凝胶、水、聚乙二醇、丙二醇、乙二醇、蓖麻油或氢化蓖麻油或多乙氧基氢化蓖麻油、芝麻油、玉米油、花生油等。
[0040]“烷基”表示1-20个碳原子的饱和的脂烃基,包括直链和支链基团(本技术书中提
到的数字范围,例如“1-20”,是指该基团,此时为烷基,可以含1个碳原子、2个碳原子、3个碳原子等,直至包括20个碳原子)。更优选的是,烷基是有1-10个碳原子的中等大小的烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基、戊基等。最好是,烷基为有1-8或1-6个碳原子的低级烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基或叔丁基等。烷基可以是取代的或未取代的。当是取代的烷基时,该取代基优选是一或多个,更优选1-3个,最优选1或2个取代基。
[0041]“羟基”表示-oh基团。
[0042]“硝基”表示-no2基团。
[0043]“氰基”表示-cn基团。
[0044]“烷氧基”表示-o-(未取代的烷基)和-o-(未取代的环烷基)。代表性实例包括但不限于甲氧基、乙氧基、丙氧基、丁氧基、环丙氧基、环丁氧基、环戊氧基、环己氧基等。
[0045]“卤素”表示氟、氯、溴或碘,优选为氟或氯。
[0046]
采用本发明的技术方案,优势如下:
[0047]
本发明提供的嘧啶酮类化合物具有良好的pak5酶活抑制作用,可以通过阻滞细胞周期,抑制肿瘤细胞增殖和迁移过程,可以显著抑制乳腺癌mcf-7细胞裸鼠移植瘤生长,因此,可以作为pak5抑制剂,应用于与pak5有关疾病的治疗中。
附图说明
[0048]
图1是划痕实验检测化合物cpd.5对乳腺癌mcf-7细胞迁移能力的影响;图1中a为划痕实验;图1中b为愈合率;
[0049]
图2是流式细胞术检测化合物cpd.5对乳腺癌mcf-7细胞周期分布的影响;在第一行和第二行中的六个图中,左侧峰代表g0/g1,右侧峰代表g2/m,中间部分代表s。
[0050]
图3是化合物cpd.5对裸鼠人乳腺癌mcf-7细胞移植瘤增殖的影响;图3中a为裸鼠人乳腺癌mcf-7细胞移植瘤组织解剖学观察;图3中b为裸鼠人乳腺癌mcf-7细胞移植瘤的瘤重变化曲线;图3中c为裸鼠人乳腺癌mcf-7细胞移植瘤的瘤体积生长曲线;
[0051]
图4是化合物cpd.5与pak5蛋白(pdb:2f57)的dock对接结果。
具体实施方式
[0052]
通过以下实施例对本发明的嘧啶酮类化合物作进一步的说明,但这些实施例不对本发明构成任何限制。
[0053]
实施例1
[0054]
1-乙基-6-(4-(甲基磺酰基)苯基)-4-(3-苯氧基苯基)-3,4,6,7-四氢-1h-吡咯[3,4-d]嘧啶-2,5-二酮(化合物cpd.5)的制备:
[0055]
第一步:反应条件为tsoh、etoh和回流
[0056][0057]
在圆底烧瓶中加入3-苯氧基苯甲醛1a(10mmol),乙基脲(12mmol),乙酰乙酸乙酯(1.26ml,10mmol)和tsoh
·
4h2o(0.24g,1mmol),加入无水乙醇搅拌,78℃加热回流。tlc监测反应。约4h后反应完成,将得到的沉淀物过滤并将粗产物从适当的溶剂中重结晶纯化,得到2.10g中间产物(4a),白色固体,收率81%。熔点为205~206℃(204~206℃);1h nmr(400mhz,cdcl3)δ8.03(s,1h),7.31-7.23(m,4h),5.72(s,1h),5.40(s,1h),4.07(dd,j1=7.2hz,j2=5.2hz,2h),2.34(s,3h),1.16(t,j=7.2hz,3h).
13
c nmr(100mhz,cdcl3)δ165.76,153.36,146.38,143.83,128.86,128.86,128.10,126.74,126.74,101.53,60.18,55.91,18.84。
[0058]
第二步:反应条件为aibn、ccl4和回流
[0059][0060]
在剧烈搅拌下将制备的1-乙基-6-甲基-2-氧代-4-(3-苯氧基苯基)-1,2,3,4-四氢嘧啶-5-羧酸乙酯中间体4a(10mmol)加入ccl4(25ml)溶液,升温至回流,1.5h内将2.67g(15mmol)nbs分次加入反应液中。继续回流10h。反应完成后,将反应冷却至室温,并过滤沉淀物。减压浓缩滤液,残余物通过柱色谱(pe:ea=5:1)法纯化,获得溴代中间体产物(5a)。
[0061]
第三步:反应条件为et3n、meoh和回流
[0062][0063]
将制备的1-乙基-6-(溴甲基)-2-氧代-4-(3-苯氧基苯基)-1,2,3,4-四氢嘧啶-5-羧酸乙酯5a(2.00mmol)和4-(甲磺酰基)苯胺(2.20mmol)溶于四氢呋喃,搅拌均匀,加入三乙胺(0.57ml,4.40mmol),加热回流约4h,反应结束。冷却至室温,过滤除去沉淀,滤液浓缩,加入乙酸乙酯(10ml
×
3)稀释,用稀盐酸、饱和碳酸氢钠和饱和食盐水(10ml
×
3)洗涤,合并
有机相,无水硫酸钠干燥,减压蒸馏获得油状粗产物,柱层析(pe:ea=8:3)获得目标化合物cpd.5。1h nmr(400mhz,dmso-d6)δ7.99-7.95(m,3h),7.88(s,1h),7.85(s,1h),7.43-7.34(m,3h),7.17-7.12(m,2h),7.05-7.00(m,3h),6.91
–
6.86(m,1h),4.95
–
4.76(m,2h),3.67-3.50(m,2h),3.17(s,3h),1.16(t,j=7.2hz,3h).
13
c nmr(100mhz,dmso-d6)δ167.80,157.18,156.72,153.00,151.53,145.67,144.32,134.11,130.60,130.51,130.51,128.60,128.60,124.06,121.85,121.85,119.16,119.16,117.93,117.41,117.08,104.28,52.66,47.77,44.33,38.55,14.63.ir(kbr):3388,3101,2924,1770;hrms(esi)calcd for:c
27h25
n3o5sna[m+na]
+
:526.1413,found:526.1390.
[0064]
实施例2化合物cpd.1的制备
[0065]
化合物cpd.1的具体实验过程参照实施例1,其合成路线如下:
[0066][0067]
参照实施例1中的制备方法,得到化合物cpd.1,白色固体产物,0.512g,收率65.07%。1h nmr(400mhz,dmso-d6)δ9.38(s,1h),7.87(s,1h),7.38(q,j=7.6hz,3h),7.30(s,1h),7.17
–
7.05(m,4h),7.01(d,j=7.7hz,3h),6.88(d,j=8.0hz,1h),6.45(d,j=7.4hz,1h),5.24(s,1h),4.71(m,2h),3.07(s,1h),1.14(t,j=7.2hz,3h).
13
c nmr(100mhz,dmso-d6)δ167.37,158.18,157.14,156.74,151.77,151.64,145.95,141.16,130.50,130.50,129.89,129.89,124.03,121.80,119.14,119.14,117.82,117.02,110.26,108.76,105.48,104.79,52.72,47.75,27.30,14.59.ir(kbr):3450(nh),3103,2980,1728,1232;hrms(esi)calcd for c
26h23
n3o4na[m+na]
+
:464.1586,found:464.1564.
[0068]
实施例3化合物cpd.2的制备
[0069]
化合物cpd.2的具体实验过程参照实施例1,其合成路线如下:
[0070][0071]
参照实施例1中的制备方法,得到化合物cpd.2,收率67%。1h nmr(400mhz,dmso-d6)δ8.13(s,1h),7.79(d,j=2.7hz,1h),7.53(dd,j=3.2,5.6hz,2h),7.42-7.35(m,2h),7.32-7.23(m,2h),7.20
–
7.06(m,3h),7.03-6.95(m,2h),6.87(s,1h),5.22(s,1h),4.45(m,2h),4.14(m,2h),4.04(s,2h),1.03(t,j=7.4hz,3h).
13
c nmr(100mhz,dmso-d6)δ168.67,161.36,157.14,156.76,151.91,151.72,146.13,134.69,131.79,131.70,130.54,130.48,130.06,129.98,124.01,121.72,119.11,117.85,116.84,116.02,115.87,115.81,115.66,104.11,52.80,47.20,42.04,38.20,14.55.ir(kbr):3390,3169,2972,1722;hrms(esi)calcd for c
27h24
fn3o3na[m+na]
+
:480.1699,found:480.1708.
[0072]
实施例4化合物cpd.3的制备
[0073]
化合物cpd.3的具体实验过程参照实施例1,其合成路线如下:
[0074][0075]
参照实施例1中的制备方法,得到化合物cpd.3,收率54%。1h nmr(400mhz,dmso-d6)δ7.87(d,j=1.8hz,1h),7.65(d,j=8.8hz,1h),7.35(dd,j=7.6,6.8hz,1h),7.23(d,j=8.8hz,1h),7.12(t,j=7.3hz,1h),7.04
–
6.96(m,1h),6.86(dd,j=8.0,1.8hz,1h),5.23(s,1h),4.79-4.65(m,2h),3.34(s,2h),2.42(s,3h),1.13(t,j=6.8hz,3h).
13
c nmr(100mhz,dmso-d6)δ167.34,157.17,156.74,151.76,151.74,145.91,137.72,131.57,131.57,130.55,130.49,130.49,127.80,124.03,121.81,121.81,119.16,118.83,118.83,117.84,117.04,104.67,52.75,47.68,38.42,16.16,14.62.ir(kbr):3332,3153,2978,1732;hrms(esi)calcd for:c
27h25
n3o3sna[m+na]
+
:494.1514,found:494.1523.
[0076]
实施例5化合物cpd.4的制备
[0077]
化合物cpd.4的具体实验过程参照实施例1,其合成路线如下:
[0078][0079]
参照实施例1中的制备方法,得到化合物cpd.4,白色固体产物,0.512g,收率65.07%。1h nmr(400mhz,cdcl3)δ8.57(s,1h),7.28(d,j=4.0hz,4h),7.25(dd,j=8.0,4.0hz,1h),6.80(d,j=8.0hz,1h),6.74(s,1h),6.71(s,1h),6.03(s,1h),5.37(s,1h),3.97(s,2h),3.86(s,3h),3.85(s,3h),2.84(t,j=6.0hz,2h),2.75(t,j=6.6hz,2h).
13
c nmr(100mhz,cdcl3)δ165.38,152.64,149.03,148.60,147.61,143.96,131.71,128.64,128.64,127.85,126.56,126.56,120.60,111.92,111.43,99.52,59.94,55.94,55.87,51.12,47.70,35.73.ir(kbr,cm-1
):v 3217,3063,2932,2832,1690,1543,1512,1458,1389,1312,1265,1234.ms(esi)calcd for c
22h23
n3o
6 394.2[m+h]
+
,found:394.8.
[0080]
以下是本发明的代表化合物的部分药理试验及结果:
[0081]
1、cck-8法测试肿瘤细胞的增殖抑制活性
[0082]
胰酶消化培养皿中处于对数生长期的细胞,计数板计数,96孔板除周边孔只加200μl pbs外,其余每孔铺3000-6000个细胞;设置4个复孔,每孔加10%fbs的完全培养基100μl;细胞贴壁后,倒扣弃去96孔板中培养基,加入含有不同浓度的化合物的培养基,置37℃,5%co2培养箱中培养48h;48h后弃去96孔板中旧培养基,每孔加入90μl无血清培养基,10μl cck-8溶液,混匀,避光操作;放入培养箱中继续孵育0.5,1,2,4h;酶标仪450nm测定od值。实验重复三次。相对抑制率=(od
对照组-od
实验组
)/od
对照组
×
100%。
[0083]
表1显示本发明代表化合物1-5对于四种肿瘤细胞(achn(人肾癌细胞)、hepg2(人肝癌细胞)、hct116(人结肠癌细胞)和mcf-7(人乳腺癌细胞)的活性数据。活性使用ic
50
表征。以5-氟尿嘧啶(5-fu)和嘧啶酮母核化合物作为阳性对照,其中,嘧啶酮母核化合物的结构式如下:
[0084][0085][0086]
由表1可知,本技术代表化合物对体外培养的肿瘤细胞achn、mcf-7、hct-116和hepg2具有良好抗肿瘤活性,其他化合物也具有类似的效果。其中,化合物cpd.5对乳腺癌细胞mcf-7具有良好的增殖抑制活性(ic
50
=2.7
±
0.1μm),抗肿瘤活性最佳。
[0087]
2、htrf试剂盒检测pak5酶活抑制作用
[0088]
使用均相时间分辨荧光(htrf)stk-s2试剂盒(62st2peb,cisbio)确定pak5抑制活性。pak5蛋白购自abcam。简而言之,激酶反应在96孔微孔板(cisbio)中进行,每孔反应体积为10μl,其中包含指定浓度的待测化合物,5μm肽底物,1.5ng/μl pak5和500μm atp在激酶缓冲液中。在室温下孵育20分钟后,添加5μl链霉亲和素-xl665和5μl stk-抗体的检测缓冲液来终止反应。将板密封并在室温下孵育1小时,然后在envision-perkinelmer上测量得到的trf能量转移信号。在620nm(抗体)和665nm(xl665)处测量荧光发射。计算每个孔的发射率(665/620),并根据下式计算每种化合物浓度的抑制百分比:抑制百分比=(max-test)/(max-min)
×
100(“max”表示无化合物对照的比例,“最小”表示无激酶对照的比例。ic
50
值使用graphpad prism 7进行测定。
[0089]
表2显示本发明代表化合物cpd.1-cpd.5对于pak5的活性数据,活性使用ic
50
表征。
[0090][0091]
采用htrf kinease试剂盒检测pak5激酶活性,由表2可知,本技术代表化合物具有良好的pak5抑制活性,其他化合物也具有类似的效果。其中,化合物cpd.5的酶活抑制能力超过泛pak抑制剂舒尼替尼,活性效果最佳。
[0092]
3、化合物cpd.5抑制乳腺癌细胞mcf-7迁移
[0093]
使用划痕实验,研究化合物cpd.5对肿瘤细胞迁移的影响,实验结果见图1。分别用1μm和5μm的化合物cpd.5和阳性对照药5-fu处理mcf-7细胞,在0h和24h拍摄各组mcf-7细胞迁移的情况。
[0094]
实验结果表明:5-fu组和化合物cpd.组处理的mcf-7细胞在24h时的迁移率均比空白对照control组低,并且随着给药浓度的增加,其抑制细胞迁移的能力进一步加强,mcf-7细胞的迁移率逐渐降低,甚至优于对照药5-fu。与对照组相比差异有统计学意义(****p《0.0001)。浓度为5μm时,化合物cpd.5组mcf-7细胞的迁移率明显比5-fu低,说明化合物cpd.5能够抑制乳腺癌细胞mcf-7的迁移,差异具有统计学意义(****p《0.0001)。
[0095]
4、化合物cpd.5诱导乳腺癌细胞mcf-7周期阻滞
[0096]
mcf-7细胞经不同浓度的化合物cpd.5(0.1、0.5、1μm)、嘧啶酮母核和5-氟尿嘧啶处理36h后,用pi单染结合流式细胞术检测细胞周期,直方图显示细胞周期分布百分比。
[0097]
由图2可知,空白对照组mcf-7细胞大部分位于g0/g1期,经嘧啶酮母核、低浓度(0.1-0.5μm)化合物cpd.5作用后,各细胞周期细胞比例变化不大(p》0.05)。高浓度(1μm)化合物cpd.5作用后,相对空白对照组,g0/g1期细胞比例明显降低,g2/m期细胞比例增加(p《0.05),说明化合物cpd.5诱导mcf-7细胞的g2/m期阻滞。
[0098]
5、化合物cpd.5在体内抗肿瘤活性研究
[0099]
使用4-6周龄雌性spf级balb/ca-nu裸鼠建立mcf-7裸鼠移植瘤模型,30天左右开始成瘤,瘤体积达到100mm3后,随机分成四组开始给药,连续观察20天,隔天称量裸鼠体重以及成瘤体积,20天后将小鼠处死,摘除肿瘤(图3a)。
[0100]
在用药期间,小鼠未见死亡与不良症状,与空白对照组相比,化合物cpd.5给药组的裸鼠体重无明显变化,表明化合物在体内没有毒性。在同等给药剂量条件下,化合物cpd.5组的抑瘤率达到52.8%,比阳性对照药组5-fu提高了6.9%,体现了化合物cpd.5具有良好的体内抗肿瘤活性。
[0101]
6、分子对接结果
[0102]
化合物cpd.5与pak5蛋白的分子对接结果如图4所示:化合物cpd.5可以同时与
pak5蛋白的铰链区和dfg区相互作用。其中,4-苯基-3,4,6,7-四氢-1h-吡咯并[3,4-d]嘧啶-2,5-二酮结构中2位的羰基可以与铰链区关键氨基酸残基leu526形成氢键;6位侧链上的甲磺酰基可以和dfg区的asp586形成氢键,从而抑制pak5的酶活。
[0103]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1