一种酶促反应结晶制备高纯度头孢克洛的方法
1.本发明属于药物结晶领域,具体涉及一种酶促反应结晶制备高纯度头孢克洛的方法。
背景技术:
2.头孢克洛属于第二代口服头孢菌素类抗生素药物,对多种革兰氏阳性菌和革兰氏阴性菌均具有很强的杀灭作用,因具有广谱、高效和良好的临床安全性,已成为目前治疗细菌感染的重要药物之一。临床上主要用于皮肤及软组织感染(皮下脓疡、毛囊炎),呼吸道感染(如咽炎、肺炎),尿路感染(如肾盂肾炎、膀胱炎)。头孢克洛在医生处方量与零售药店销售额方面均居世界抗菌药物首位,多年来一直是全球畅销的口服头孢类抗生素。
3.头孢克洛在工业上的制备方法主要有化学合成法和生物酶法。现有化学合成法存在工艺路线复杂、成本费用高、收率低、污染大等问题。生物酶法不仅可以减少反应步骤,还具有环境友好、效率高、生产成本低等优点。由于头孢克洛在青霉素g酰化酶(pga)存在时易发生水解反应而分解,即产物不稳定,因此通过结晶的方式将其从液相移除,对整个反应的进行有积极影响。
4.目前,在生物酶法制备头孢克洛的现有报道中,尽管也有研究者采用酶促反应和结晶联合的策略,但大多都先进行酶促反应,反应结束后得到液相粗品,再对粗品进行结晶提纯。该方法没有从根本上解决pga催化的头孢克洛合成反应中产物分解的问题,所以反应步骤的效率较低。因此,研制一种酶促反应结晶一步制备头孢克洛晶体产品的方法,对头孢克洛的高效生产及广泛应用具有十分重要的意义。
技术实现要素:
5.本发明的目的在于克服现有技术的不足,提供一种酶促反应结晶一锅制备高纯度头孢克洛的方法。该方法在酶促反应进行的同时,选择合适的过饱和度加入适量头孢克洛晶种诱导产物结晶,并进行反应结晶调控,最终可以直接得到高纯头孢克洛晶体。该方法在解决酶法生产过程产物分解问题的同时,实现了酶促反应结晶一步制备出高纯度头孢克洛(纯度99%以上)。
6.为解决上述技术问题,本发明采用如下技术方案:
7.一种酶促反应结晶制备高纯度头孢克洛的方法,该方法包括如下步骤:
8.步骤(1):将7-氨基-3-氯-头孢烯酸(7-acca)和d-苯甘氨酸甲酯盐酸盐(pgme)加入到浓度为0.1~0.3mol/l、ph为6.0~6.5的磷酸盐缓冲液中,20~25℃、磁力搅拌下,加入青霉素g酰化酶(pga),进行酶催化合成反应;
9.其中,每1.0~1.5克7-氨基-3-氯-头孢烯酸加15~25毫升磷酸盐缓冲液;7-氨基-3-氯-头孢烯酸和d-苯甘氨酸甲酯盐酸盐的摩尔比为1:1.2~1:1.6;每mmol7-氨基-3-氯-头孢烯酸加入220~300u青霉素g酰化酶;
10.步骤(2):当酶催化合成反应液中的头孢克洛的过饱和度为2.0~2.4时,再加入头
孢克洛晶种进行反应结晶,搅拌下反应140~200min,反应结束后进行抽滤、干燥,得到头孢克洛产品;
11.其中,每ml酶催化合成反应液加入0.004~0.006g的头孢克洛晶种。
12.所述的步骤(1)、(2)中酶催化反应中的搅拌速率为150~200r/min。
13.所述的头孢克洛晶种的纯度为99%。
14.步骤(2)中,在添加晶种后的搅拌反应过程中,还加入d-苯甘氨酸甲酯盐酸盐溶液,d-苯甘氨酸甲酯盐酸盐溶液的浓度为80~120mmol/l,添加的d-苯甘氨酸甲酯盐酸盐的质量是步骤(1)中7-氨基-3-氯-头孢烯酸质量的10~20%。
15.本发明的实质性特点为:
16.现有制备头孢克洛的方法是在酶促反应结束后,要对所得头孢克洛料液粗品进行结晶提纯,再经过滤、干燥,才能得到头孢克洛产品。
17.而本发明的实验步骤是在酶促反应过程中进行反应结晶调控后,直接进行过滤、干燥就可以得到纯度99%以上的头孢克洛产品,省却了对粗品的结晶提纯步骤。
18.本发明所用方法不同,在酶促反应进行的同时,选择合适的过饱和度加入适量头孢克洛晶种诱导产物结晶,并进行反应结晶调控,最终直接得到高纯头孢克洛晶体。本发明通过酶促反应和结晶的高效耦合,在解决酶法生产过程产物分解问题的同时,实现了一锅直接得到头孢克洛晶体产品。
19.本发明的有益效果是:
20.本发明的酶促反应结晶法制备头孢克洛,通过将结晶产物移除技术耦合到酶促反应过程,并进行全程调控,最终得到高纯度头孢克洛产品。该方法不需要使用任何有机溶剂,绿色无污染,反应条件温和,能耗低;不需要控制反应过程的ph值,简便易控;在酶促反应过程中进行结晶调控,直接一步从反应原料得到纯度合格的头孢克洛晶体产品(99%以上,最高可达99.5%),工艺路线短,简化了分离纯化的步骤,以工业化产量5000g来计,仅从避免粗品结晶的步骤就能节省1.5万元费用,为工业化提供一种高效率的生产方法。
21.此外,本发明操作简单,不需要反应过程中控制溶液的ph值(因为当前技术中,头孢克洛制备过程中大部分都需要用ph自动滴加仪维持整个制备过程中的ph值保持不变,)简便易控;制备过程中不需要使用任何有机溶剂,绿色无污染,反应条件温和,能耗低。
附图说明
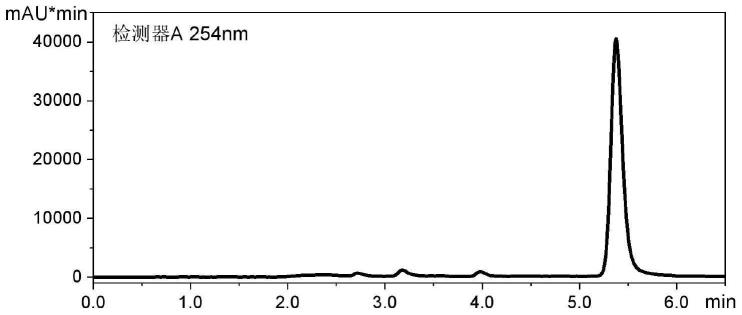
22.图1为实施例1制备得到的头孢克洛产品的高效液相色谱图。
23.图2为实施例1制备得到的头孢克洛产品的显微镜图片。
具体实施方式
24.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。具体实施例仅是对本发明的一种说明,而不构成对本发明的限制。下述实施例中所用的试验方法如无特殊说明,均为常规方法;所使用的各种原材料、试剂、仪器和设备等,如无特殊说明,均可通过市场购买得到或者可通过现有方法制备得到。
25.实施例1:
26.在反应器中加入7-氨基-3-氯-头孢烯酸1.23g(5.24mmol)、d-苯甘氨酸甲酯盐酸
盐1.587g(7.87mmol)、0.2mol/l的磷酸盐缓冲液(ph=6.5)20ml,在150r/min的磁力搅拌下,向体系中滴加氨水(质量浓度25~28%)至7-acca完全溶解,再用盐酸(浓度36~38%)缓慢调节其ph至6.5,最后用磷酸盐缓冲液调整体积至25ml,从而得到7-acca的过饱和溶液体系。用超级恒温水浴将结晶器温度控制在20℃,准确吸取1.26ml酶液(游离酶:青霉素g酰化酶,这里游离酶的酶活是1000u/ml。)加入到反应体系中,立即发生酶催化反应。反应进行5min时(在制备过程中都需要用高效液相色谱仪检测头孢克洛的浓度,用液相色谱检测在反应进行5min时头孢克洛的饱和度为2.0~2.4)加入0.145g头孢克洛晶种(纯度为99%),进行酶促反应结晶过程。180min后停止搅拌,抽滤,将滤饼在0.085mpa、40℃下干燥4h,得到头孢克洛产品,高效液相色谱仪测得其纯度为99.5%。
27.头孢克洛产品的高效液相色谱图如图1所示,副产物苯甘氨酸的保留时间是3.178min,产品头孢克洛的保留时间是5.376min,根据出峰面积与样品浓度的关系计算出头孢克洛产品的纯度为99.5%;头孢克洛产品的显微镜图片如图2所示,其头孢克洛产品的晶习为块状。
28.实施例2:
29.在反应器中加入7-氨基-3-氯-头孢烯酸1.23g(5.24mmol)、d-苯甘氨酸甲酯盐酸盐1.587g(7.87mmol)、0.2mol/l的磷酸盐缓冲液(ph=6.5)20ml,在150r/min的磁力搅拌下,向体系中滴加氨水(质量浓度25~28%)至7-acca完全溶解,再用盐酸(浓度36~38%)缓慢调节其ph至6.5,最后用磷酸盐缓冲液调整体积至25ml,从而得到7-acca的过饱和溶液体系。用超级恒温水浴将结晶器温度控制在20℃,准确吸取1.26ml酶液(游离酶:青霉素g酰化酶,这里游离酶的酶活是1000u/ml。)加入到反应体系中,立即发生酶催化反应。反应进行5min时(在制备过程中都需要用高效液相色谱仪检测头孢克洛的浓度,用液相色谱检测在反应进行5min时头孢克洛的饱和度为2.0~2.4)加入0.145g头孢克洛晶种,进行酶促反应结晶过程,反应进行60min时流加浓度为100mmol/l的d-苯甘氨酸甲酯盐酸盐溶液,流加时间为30min,流加总量为10ml。180min后停止搅拌,抽滤,将滤饼在0.085mpa、40℃下干燥4h,得到头孢克洛产品,高效液相色谱仪测得其纯度为99.1%。本实施例通过d-苯甘氨酸甲酯盐酸盐溶液的加入,在保证纯度的基础上来提高产率。
30.实施例3:
31.在反应器中加入7-氨基-3-氯-头孢烯酸1.23g(5.24mmol)、d-苯甘氨酸甲酯盐酸盐1.482g(7.34mmol)、0.2mol/l的磷酸盐缓冲液(ph=6.5)20ml,在150r/min的磁力搅拌下,向体系中滴加氨水(质量浓度25~28%)至7-acca完全溶解,再用盐酸(浓度36~38%)缓慢调节其ph至6.5,最后用磷酸盐缓冲液调整体积至25ml,从而得到7-acca的过饱和溶液体系。用超级恒温水浴将结晶器温度控制在22℃,准确吸取1.26ml酶液(游离酶:青霉素g酰化酶,这里游离酶的酶活是1000u/ml。)加入到反应体系中,立即发生酶催化反应。反应进行8min时(在制备过程中都需要用高效液相色谱仪检测头孢克洛的浓度,用液相色谱检测在反应进行8min时头孢克洛的饱和度为2.0~2.4)加入0.145g头孢克洛晶种,进行酶促反应结晶过程。200min后停止搅拌,抽滤,将滤饼在0.085mpa、40℃下干燥4h,得到头孢克洛产品,高效液相色谱仪测得其纯度为99.4%。
32.实施例4:
33.在反应器中加入7-氨基-3-氯-头孢烯酸1.23g(5.24mmol)、d-苯甘氨酸甲酯盐酸
盐1.27g(6.29mmol)、0.2mol/l的磷酸盐缓冲液(ph=6.5)20ml,在200r/min的磁力搅拌下,向体系中滴加氨水(质量浓度25~28%)至7-acca完全溶解,再用盐酸(浓度36~38%)缓慢调节其ph至6.5,最后用磷酸盐缓冲液调整体积至25ml,从而得到7-acca的过饱和溶液体系。用超级恒温水浴将结晶器温度控制在20℃,准确吸取1.26ml酶液(游离酶:青霉素g酰化酶,这里游离酶的酶活是1000u/ml。)加入到反应体系中,立即发生酶催化反应。反应进行10min时(在制备过程中都需要用高效液相色谱仪检测头孢克洛的浓度,用液相色谱检测在反应进行10min时头孢克洛的饱和度为2.0~2.4)加入0.145g头孢克洛晶种,进行酶促反应结晶过程。200min后停止搅拌,抽滤,将滤饼在0.085mpa、40℃下干燥4h,得到头孢克洛产品,高效液相色谱仪测得其纯度为99.5%。
34.对比例1:
35.其他步骤同实施例1,不同之处为,其中d-苯甘氨酸甲酯盐酸盐的加入量替换为2.12g(10.51mmol)。
36.得到的头孢克洛产品纯度为78%。由于增加了d-苯甘氨酸甲酯盐酸盐的添加量,促进了d-苯甘氨酸甲酯盐酸盐的水解反应,副产物d-苯甘氨酸的生成量增多,导致头孢克洛产品的纯度降低。
37.对比例2:
38.其他步骤同实施例1,不同之处为,其中加入0.2g的头孢克洛作为晶种。
39.得到的头孢克洛产品纯度为98.8%。由于晶种的添加量过大,过浓的晶种会加大反应体系的粘度,而且容易发生聚结现象,不利于产物结晶,导致头孢克洛产品的纯度降低。
40.对比例3:
41.其他步骤同实施例1,不同之处为,其中在反应进行10min时(过饱和度为2.9)加入0.145g头孢克洛晶种。
42.得到的头孢克洛产品纯度为98.6%。由于过饱和度较高,可能发生了爆发成核,对结晶过程不利,导致头孢克洛产品的纯度降低。
43.通过以上实施例和对比例说明,本发明的方法在酶促反应结晶过程中晶种浓度的选择,晶种浓度太低,不足以提供足够量的结晶核心,反应周期会较长;晶种浓度太高,会加大反应体系的粘度,还可能会发生聚结现象,不利于产物结晶,并且浓度太低或过高都会降低产品纯度。
44.上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围,凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
45.本发明未尽事宜为公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1