1.本发明属于医药领域,具体涉及一种艾沙康唑中间体的制备方法。
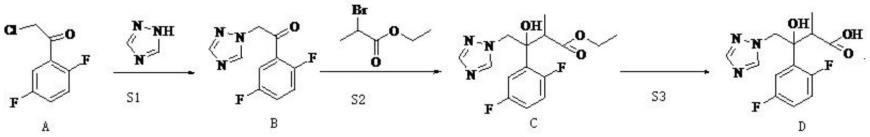
背景技术:2.艾沙康唑,cas:241479-67-4,是一款抗真菌感染药物。而如化合物f所示是合成艾沙康唑的重要中间体。化合物f的合成路线如下式所示:
[0003][0004]
在上述合成路线中,由于催化剂的限制,s1步骤通常收率较低。
技术实现要素:[0005]
为了解决上述问题,本技术提供一种艾沙康唑中间体的制备方法。本发明通过提供cui掺杂纳米微球nio催化剂,以提高该步骤的收率。
[0006]
为了实现上述目的,本发明采用如下技术方案:
[0007]
一种艾沙康唑中间体的制备方法,所述艾沙康唑中间体的结构式如化合物f所示,化合物f的合成路线如下式所示,
[0008][0009]
s1中,所用催化剂为cui掺杂纳米微球nio催化剂,所述纳米微球nio的粒径为200-300纳米。
[0010]
进一步的,所述cui掺杂纳米微球nio催化剂中,nio与cui的摩尔比为1:2。
[0011]
上述方案中,cui掺杂纳米微球nio催化剂,以纳米微球nio为模板,cui掺杂在纳米微球nio中,对纳米微球nio表面进行掺杂修饰,在催化化合物a与1,2,4-三氮唑的反应时,监测反应时长在5-7h内化合物a完全反应完,且经重结晶后的到化合物b收率大于85%,猜测是因为纳米微球nio的球形结构使得在催化过程中,更多的反应物与cui接触参与反应,
提高了反应效率,同时由于cui和nio的双重催化的作用使得化合物a与1,2,4-三氮唑的反应更彻底。
[0012]
进一步的,cui掺杂纳米微球nio催化剂的用量为化合物a2-5wt%。
[0013]
进一步的,所述cui掺杂纳米微球nio催化剂由以下方法制成:
[0014]
(1)将醋酸镍溶于水中制成镍盐溶液,搅拌下加入尿素,体系加热至70-80℃,反应至沉淀完全析出,然后将体系放入聚四氟乙烯不锈钢高压釜内,在180-200℃保持10-12小时,以5-6℃/min的速度冷却至室温,然后离心、纯水洗涤、乙醇洗涤,得到纳米微球nio粉末;所述尿素与醋酸镍的摩尔比为3:1;
[0015]
(2)配制硫酸铜水溶液,并在硫酸铜水溶液中加入吐温80,然后再加入所得的纳米微球nio粉末,超声10-20min,得到混合液a;配制碘化钾和硫代硫酸钠混合溶液,得到混合液b;超声条件下将混合液b加入混合液a中,析出沉淀,过滤沉淀,依次用纯水、乙醇洗涤,然后将所得沉淀真空干燥,最后将干燥后的产物放在马弗炉中煅烧1-2h,煅烧温度为200-400℃。
[0016]
上述方案中,在制备cui掺杂纳米微球nio催化剂时,步骤(1)中,氢氧化镍的生成速率、在聚四氟乙烯不锈钢高压釜内的温度,以及反应后的降温速度,都是得到目标纳米微球nio的关键控制因素,本发明采用尿素与镍盐反应,且尿素与醋酸镍的摩尔比为3:1,可避免氢氧化镍快速沉淀造成粒径不均,并控制反应釜内温度在180-200℃,且反应后的降温速度为5-6℃/min,因此得到了粒径在200-300纳米范围内的纳米微球nio,粒径大小分布符合期望目标大小,而醋酸镍的浓度影响不大,只需将醋酸镍与尿素控制在一定范围内即可;步骤(2)中,吐温80起到良好的分散作用,一方面将纳米微球nio粉末均匀的分散在体系中,另一方面,在析出cui后,可保证cui均匀分散在体系中,避免cui掺杂不均,影响催化效果,且总超声时间最好不超过30min,以免微球结构破坏影响催化效果。
[0017]
进一步的,s1具体操作为:将化合物a投入反应溶剂i中,加入cui掺杂纳米微球nio催化剂、1,2,4-三氮唑和碳酸钾,加热至温度为60-70℃,化合物a反应完全后,过滤反应体系,收集滤液,减压蒸馏浓缩滤液,然后重结晶得到化合物b;所述反应溶剂i为dmf。
[0018]
进一步的,s4:将金鸡纳碱溶于乙醇中配成溶液,再投入化合物d,加热回流反应,体系变澄清,降温析出固体,过滤收集滤饼,并用乙醇洗涤滤饼,再将滤饼溶于热乙醇中重结晶,干燥,得到羧酸盐;将该羧酸盐投入水中,用盐酸调节ph 3-4,然后用二氯甲烷萃取,有机相干燥后浓缩得到化合物e。
[0019]
进一步的,s5:室温下,将四碘化二磷加入二氯甲烷中,搅拌溶解,将la[n(sime3)2]3加入体系,再加入化合物e,搅拌反应2-5min,再加入碳酸铵,反应至化合物e完全反应,然后二氯甲烷稀释,并依次用10%亚硫酸氢钠溶液、水洗涤,然后用干燥剂干燥有机相,浓缩有机相得到粗品化合物f,最后用正庚烷/二氯甲烷混合溶剂重结晶,得到纯品化合物f;所述四碘化二磷与化合物e的摩尔比为1-1.1:1。该步反应中,选用低毒性的二氯甲烷作为反应溶剂,而二氯甲烷作为溶剂时在不加la[n(sime3)2]3的情况下化合物f重结晶后收率只能达到63%,本发明加入三硅胺基镧配合物la[n(sime3)2]3作为催化剂,化合物f重结晶后收率可达90%以上,大大提高了化合物f的收率,使得在使用低毒性的二氯甲烷作为溶剂的同时,保证了良好的收率;四碘化二磷稍过量以使得化合物e反应完全。
[0020]
与现有技术相比,本发明的有益效果在于:
[0021]
(1)本发明在s1中使用cui掺杂纳米微球nio作为催化剂,由于该催化剂的球形结构以及cui和nio的双重协同催化的作用使得化合物a与1,2,4-三氮唑的反应更快更彻底,s1反应时长在5-7h内,化合物b收率大于85%。
[0022]
(2)在制备cui掺杂纳米微球nio时,控制尿素与镍盐的用量比、控制反应釜内温度与反应后降温速度,可以稳定得到目标纳米微球nio。
[0023]
(3)s5中,选择二氯甲烷作为反应溶剂,将四碘化二磷、化合物e和碳酸铵反应,一步得到化合物f,简化了工艺步骤,另外在反应中以la[n(sime3)2]3作为催化剂,提高了以二氯甲烷作为反应溶剂时化合物e转化成化合物f的转化率,经重结晶后,纯品化合物f收率大于90%。
具体实施方式
[0024]
下面结合实施例对本发明作进一步的描述,所描述的实施例仅仅是本发明一部分实施例,并不是全部的实施例。基于本发明中的实施例,本领域的普通技术人员在没有做出创造性劳动前提下所获得的其他所有实施例,都属于本发明的保护范围。
[0025]
实施例1
[0026]
制备cui掺杂纳米微球nio催化剂:
[0027]
(1)将0.1mol醋酸镍溶于水中制成镍盐溶液,搅拌下加入0.3mol尿素,体系加热至70-80℃范围内,反应至沉淀完全析出,然后将体系放入聚四氟乙烯不锈钢高压釜内,在180-200℃保持12小时,控制冷却速度在5-6℃/min内冷却至室温,然后离心,固体依次用纯水洗涤、乙醇洗涤,得到模板纳米微球nio粉末,模板纳米微球nio的粒径为200-300纳米;
[0028]
(2)配制硫酸铜水溶液100ml,浓度为1m,并在硫酸铜水溶液中加入20mg吐温80,然后再加入所得的纳米微球nio粉末3.7g,超声10min,得到混合液a;配制碘化钾和硫代硫酸钠混合溶液100ml,碘化钾浓度为1.1m,硫代硫酸钠浓度为1.1m,得到混合液b;超声条件下将混合液b滴加入混合液a中,保证在20min内滴加完成,析出沉淀,过滤沉淀,依次用纯水、乙醇洗涤,然后将所得沉淀真空干燥,干燥温度为50℃,最后将干燥后的产物放在马弗炉中煅烧1h,煅烧温度为200℃,自然冷却得到cui掺杂纳米微球nio催化剂。
[0029]
实施例2
[0030]
s1:在将1mol化合物a投入1l dmf中,加入cui掺杂纳米微球nio催化剂3.9g、1.1mol 1,2,4-三氮唑和1.1mol碳酸钾,加热至温度为60-70℃范围内,化合物a反应完全后(反应时长约为6小时20分钟),过滤反应体系,收集滤液,减压蒸馏浓缩滤液,然后乙酸乙酯重结晶得到化合物b 191.2g,收率85.6%。
[0031]
实施例3
[0032]
s1:在将1mol化合物a投入1l dmf中,加入cui掺杂纳米微球nio催化剂9.5g、1.1mol 1,2,4-三氮唑和1.1mol碳酸钾,加热至温度为60-70℃范围内,化合物a反应完全后(反应时长约为6小时),过滤反应体系,收集滤液,减压蒸馏浓缩滤液,然后乙酸乙酯重结晶得到化合物b 194.7g,收率87.2%。
[0033]
实施例4
[0034]
s1:在将1mol化合物a投入1l dmf中,加入cui掺杂纳米微球nio催化剂3.9g、1.2mol 1,2,4-三氮唑和1.2mol碳酸钾,加热至温度为60-70℃范围内,化合物a反应完全后
(反应时长约为6小时20分钟),过滤反应体系,收集滤液,减压蒸馏浓缩滤液,然后乙酸乙酯重结晶得到化合物b 193.5g,收率86.7%。
[0035]
对比例1
[0036]
本对比例与实施例2的不同之处在于,本对比例使用cui掺杂纳米微球nio催化剂1.9g,是化合物a的1wt%,其他操作与实施例2相同。反应时长约为8小时40分钟,最终得到化合物b 177.3g,收率79.4%。
[0037]
对比例2
[0038]
本对比例与实施例2的不同之处在于,本对比例使用普通cui催化剂3.9g,其他操作与实施例2相同。反应时长约为9小时50分钟,最终得到化合物b 163.4g,收率73.2%。
[0039]
从实施例2、实施例3、对比例1和对比例2的反应时长(记录方法为,反应6小时时tlc点板测一次监测化合物a是否反应完,以后每5分钟测一次)和最终化合物b的收率来看,本发明的cui掺杂纳米微球nio催化剂的催化效果优于普通cui催化剂,具体体现在反应效率以及化合物b收率均有所提高,且不同的催化剂用量也直接影响反应效率以及最终收率,本发明将cui掺杂纳米微球nio催化剂的用量限定在化合物a 2-5wt%,既能够获得良好的收率,又能够缩短反应时长,提高反应效率。
[0040]
实施例5
[0041]
s2:惰性气体保护下,将过量锌粉投入四氢呋喃中,然后投入三甲基氯硅烷,室温下反应0.5-1h,降温至0℃,滴加1mol 2-溴丙酸乙酯,反应完成后,过滤,收集滤液;在惰性气体保护下,将0.5mol化合物b(111.6g)溶于四氢呋喃中,再将所得的化合物b溶液滴加入所得滤液中,反应完全后,过滤,收集滤液,将滤液投入乙酸乙酯,并向其中加入稀盐酸,搅拌,用水洗涤体系,收集有机相,并干燥有机相,最后浓缩有机相得到99.6g化合物c,收率为61.2%,此处所得化合物c中包含rr和ss两种构型,rs和sr两种构型已除去;
[0042]
s3:取s2所得化合物c 0.2mol(65.1g)投入乙醇,滴加氢氧化钠水溶液,反应完全后,用盐酸溶液调节ph3-4,析出固体,过滤、洗涤、干燥,得到化合物d 57.1g;
[0043]
s4:将0.2mol金鸡纳碱溶于乙醇中配成溶液,再投入0.1mol化合物d,加热回流反应,体系变澄清,即反应完全后,降温析出固体,过滤收集滤饼,并用乙醇洗涤滤饼,再将滤饼溶于热乙醇中重结晶,干燥,得到羧酸盐;将该羧酸盐投入水中,用盐酸调节ph 3-4,然后用二氯甲烷萃取,有机相干燥后浓缩得到化合物e14.2g,收率47.7%;
[0044]
s5:室温下,将0.055mol四碘化二磷加入二氯甲烷(二氯甲烷的含水量不超过0.01wt%)中,搅拌溶解四碘化二磷,将0.3g la[n(sime3)2]3加入体系中,再加入0.05mol化合物e,搅拌反应2-5min,再加入0.055mol碳酸铵,反应至化合物e完全反应,然后用二氯甲烷稀释,再依次用10%亚硫酸氢钠溶液、水洗涤,然后用干燥剂干燥有机相,浓缩有机相得到粗品化合物f,最后用正庚烷/二氯甲烷(正庚烷与二氯甲烷的体积比为1:1)混合溶剂重结晶,得到纯品化合物f12.7g,收率91.3%。
[0045]
对比例3
[0046]
本对比例与实施例5的不同之处在于反应中未加入la[n(sime3)2]3,具体如下:
[0047]
s5:室温下,将0.055mol四碘化二磷加入二氯甲烷(含水量不超过0.01wt%)中,搅拌溶解四碘化二磷,再加入0.05mol化合物e,搅拌反应2-5min,再加入0.055mol碳酸铵,反应至化合物e完全反应,然后二氯甲烷稀释,并依次用10%亚硫酸氢钠溶液、水洗涤,然后用
干燥剂干燥有机相,浓缩有机相得到粗品化合物f,最后用正庚烷/二氯甲烷(正庚烷与二氯甲烷的体积比为1:1)混合溶剂重结晶,得到纯品化合物f 8.8g,收率63%。
[0048]
从实施例5和对比例3的数据可知,反应中未加入la[n(sime3)2]3最终收率远小于添加la[n(sime3)2]3的收率。