一种Apaf1基因敲除HEK293细胞系及构建方法和应用
一种apaf1基因敲除hek293细胞系及构建方法和应用
技术领域
1.本发明涉及一种apaf1基因敲除hek293细胞系及构建方法和应用,属于分子生物学技术领域。
背景技术:
2.重组蛋白药物是指应用重组dna或rna技术获得的治疗性的蛋白质,在目前生物制药领域占据重要地位。目前在工业生产中,哺乳动物细胞是重组蛋白类药物最常用的表达系统之一,中国仓鼠卵巢(chinese hamster ovary,cho)细胞是目前重组蛋白质生产的首选宿主细胞,用于治疗的重组蛋白药物约70%是由cho细胞制备,但在cho细胞中生产重组蛋白的一个缺点是缺乏正确的翻译后糖基化修饰功能,这会促使重组蛋白在人体内具有免疫原性。而hek293细胞作为人源细胞种系,具有精确的糖基化模式,消除了免疫原性的可能性,可作为重组蛋白生产的替代宿主。但是随着人们的需求量增加,进一步提高重组蛋白药物产量、降低生产成本成为亟待解决的问题。
3.目前,提高重组蛋白药物产量的措施涉及重组蛋白载体优化,细胞系建立以及培养基及生产工艺等的改进。伴随着基因工程尤其是基因编辑技术的进步,改造细胞系这一手段在提高重组蛋白产量上被越来越广泛的应用。
4.在众多的研究中,我们注意到抑制细胞凋亡途径在提高重组蛋白产量上具有极大潜力,通过抑制细胞凋亡途径,可以减缓细胞凋亡,增加细胞数量,从而提高重组蛋白产量。其中apaf1(凋亡酶激活因子,apoptotic protease activating factor-1)是一种分子量为130kda的蛋白质,在依赖线粒体凋亡途径中发挥着重要作用。它包括一个n-末端的card结构域,接着是一个与ced-4高度同源的区域和一个包含多个wd-40重复序列的c-末端结构域,它在细胞色素c和datp存在的情况下寡聚形成一个非常大的(~700-1400kda)的凋亡体复合体,该复合体与procaspase-9结合并催化其活性,激活的procaspase-9通过级联反应激活下游成分,促进细胞凋亡。因此,通过抑制apaf1减缓细胞凋亡途径,进而延长宿主细胞表达时限,使提高重组蛋白生产成为可能。目前,apaf1基因敲除的hek293细胞系及其在重组蛋白表达方面未见报道。
技术实现要素:
5.为了弥补现有技术中的不足,本发明的第一个目的是提供一种apaf1基因敲除hek293细胞系,从基因和蛋白水平的检测都证实,apaf1基因已被成功敲除。
6.本发明的第二个目的是提供的apaf1基因敲除hek293细胞系构建方法,该方法基于crispr/cas9技术,可以简单、高效的实现hek293细胞中apaf1基因的敲除。与沉默、敲低、干扰等方法相比,敲除效果更加彻底,适于对apaf1基因缺失的hek293细胞进行更加深入的研究。
7.本发明的第三个目的是提供apaf1基因敲除hek293细胞系在重组蛋白生产中的应用。
8.为了实现上述目的,本发明所采用的技术方案是:
9.一种apaf1基因敲除hek293细胞系,对hek293细胞中的apaf1基因进行敲除,得到apaf1基因敲除的hek293细胞系。
10.本发明提供的apaf1基因敲除hek293细胞系,与正常的hek293细胞系相比,凋亡水平受到抑制,增殖较为明显。使用apaf1基因敲除hek293细胞瞬时或稳定表达重组蛋白,与正常hek293细胞相比,重组蛋白表达量显著提高。
11.优选地,所述敲除为利用crispr/cas9技术实现。
12.crispr/cas9技术相对于锌指核酸酶(zfn)、转录激活因子样效应分子(talen),具有更高效、快速、简便、廉价的优点,利用crispr/cas9技术可以快速、高效、低成本地完成hek293细胞中apaf1基因的敲除。
13.优选地,所述hek293细胞为hek293e细胞。
14.hek293e细胞转染效率高,易于sgrna的转染,且易于培养,便于后期单克隆细胞的获得。
15.一种apaf1基因敲除hek293细胞系构建方法,包括以下步骤:根据人apaf1基因序列设计sgrna,将其分别插入到表达载体中构建sgrna表达载体;sgrna表达载体转染hek293细胞,经嘌呤霉素筛选、细胞单克隆化处理、鉴定后获得敲除apaf1基因的hek293细胞系。
16.本发明提供的apaf1基因敲除hek293细胞系构建方法,能快速高效的实现hek293细胞中apaf1基因的敲除,且便于操作,成本较低。
17.优选地,所述sgrna的核苷酸序列如seq id no1~3所示。
18.本发明依据人apaf1基因中5个转录本的共有外显子序列进行sgrna的设计,上述三个sgrna序列可对apaf1基因进行高效敲除。
19.优选地,所述sgrna的敲除靶点位于apaf1基因第三外显子上;所述apaf1基因第三外显子的核苷酸序列如seq id no5所示。
20.优选地,所述嘌呤霉素的浓度为1.0~1.5μg/ml。
21.使用此浓度范围的嘌呤霉素对转染sgrna表达载体的hek293细胞进行筛选,可以在保证转染成功的hek293细胞存活的同时,杀死没有成功转染的hek293细胞,且细胞的生长状态没有影响。
22.apaf1基因敲除hek293细胞系在重组蛋白生产中的应用。
23.与正常hek293细胞相比,在apaf1基因敲除hek293细胞中瞬时或稳定表达重组蛋白,重组蛋白的表达量显著升高。apaf1基因敲除hek293细胞有望为重组蛋白的高效表达与生产提供一种有前景的宿主细胞。
附图说明
24.图1为本发明实施例1中sgrna表达载体图;
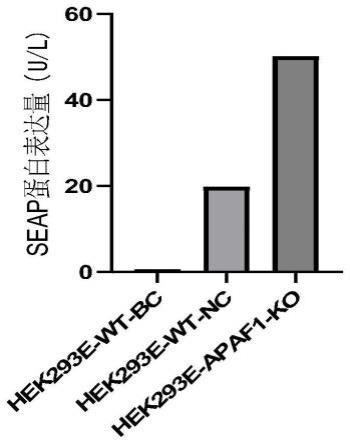
25.图2为本发明实施例1中sgrna表达载体转染效率荧光图(40倍);
26.图3为本发明实施例1中有限稀释法获得apaf1基因敲除hek293单克隆细胞荧光图(100倍);
27.图4为本发明实施例1中获得apaf1基因敲除hek293单克隆细基因测序结果;
28.图5为本发明实施例1中敲除细胞系apaf1的mrna表达水平、蛋白表达水平检测结
果;
29.图6为本发明实施例1中敲除细胞系悬浮培养7天的细胞计数结果;
30.图7为本发明实施例2中本发明中seap蛋白检测结果;
31.图8为本发明实施例2中本发明中vn蛋白的western blot表达水平检测结果(m为marker)。
具体实施方式
32.下面结合具体附图和实施例对本发明进行详细、明确的说明,但是下述说明仅是本发明的较佳实施方式而已,不用来限定本发明的保护范围。
33.下述实施例中若未特别指明,所用材料、试剂等,均从商业途径得到,所用技术手段为本领域技术人员所熟知的常规手段。下述实施例中的试验方法,如无特别说明,均为常规方法。
34.实施例1一种apaf1基因敲除hek293细胞系及构建方法
35.本实施例的apaf1基因敲除hek293细胞系基于crispr/cas9技术对hek293细胞中的apaf1基因进行敲除,得到apaf1基因敲除的hek293细胞系,具体实施操作如下:
36.1.靶点引物设计
37.以人源基因组序列为目标,在(http://www.ncbi.nlm.nih.gov)查询apaf1基因组序列,可见其mrna基因组序列有5个转录本,通过序列对比,可见转录本1具有其他转录本所有外显子基因序列,故选择转录本1作为设计sgrna的模板,转录本1的基因序列如seq id no.4所示。我们选择5个转录本的共有外显子序列进行sgrna的设计,以atg下游的第一个外显子,以期增加移码突变机率,即mrna基因组序列的第三个外显子为敲除靶点,其序列如seq id no.5。利用在线设计网站(crispor.tefor.net)对apaf1基因进行靶点设计,根据网站对sgrna的评分高低,我们选择评分较高的前三个序列,分别为命名为sgrna1、sgrna2、sgrna3,核苷酸序列如seq id no.1~3所示。
38.2.敲除载体构建
39.委托天津微思远生物科技公司进行载体构建(sgrna表达载体的出发载体购自天津微思远生物科技公司),共构建三组敲除载体:yko-rp003-hapaf1-1、yko-rp003-hapaf1-2、yko-rp003-hapaf1-3载体图谱如图1所示(天津微思远生物科技公司提供)。
40.3.嘌呤霉素筛选
41.yko-rp003载体具有嘌呤霉素抗性,可用于后期阳性克隆细胞筛选。
42.需要确定嘌呤霉素对正常细胞的最低致死量,不同哺乳动物细胞对嘌呤霉素的敏感性和耐受浓度不同,致死剂量大概范围在0.5μg/ml-10μg/ml之间,参考其他文献所用嘌呤霉素剂量,将hek293e细胞按20万/ml的密度铺板至12孔板,覆盖率达80%时,添加不同剂量的嘌呤霉素(0.5、0.8、1.0、1.2、1.5、1.8μg/ml),维持培养5-7日,观察可引起正常细胞死亡的最低剂量。
43.将hek293e细胞在6孔板进行铺板,铺板密度按50万/ml,待细胞覆盖密度达70-80%时,将构建好的基因编辑质粒转染细胞,转染剂量2μg/ml,6h后进行换液,继续培养,转染48h后拍荧光检测转染效率,如图2所示,更换为嘌呤霉素浓度为1.2μg/ml培养基进行培养,每隔2-3日更换培养基,筛选3周后得到cell pool。
44.4.获得单克隆细胞株
45.将筛选获得的cell pools细胞更换为不含嘌呤霉素的培养基进行扩大培养,利用有限稀释法进行单克隆细胞的筛选,具体实施如下:将筛选细胞经胰酶消化后,培养基重悬,进行计数,取含有1500个细胞的培养基悬液,移至15ml离心管,用培养基稀释至10ml,枪头吹匀,按100μl分别加入96孔板,保证每孔有至少一个细胞,随后每日观察每孔细胞一周左右,单个细胞会出现典型的聚集生长,如图3所示,对单个细胞生长的孔进行标记。96孔板细胞生长速率不一,及时观察细胞,将达到传代覆盖率的细胞转移至24孔板或12孔板,持续扩大培养单克隆细胞,最终转至6孔板,及时进行细胞冻存,防止污染。
46.将扩大的单克隆细胞进行收集,利用dna提取试剂盒进行dna基因组提取,设计包含敲除位点(apaf1基因组第三外显子)在内的大小约668bp基因组片段的上下游引物,序列如seq id no.6、seq id no.7所示,进行pcr扩增,反应体系如下:
[0047][0048]
对扩增所得产物进行琼脂糖核酸电泳跑胶,将668bp大小的目的片段进行凝胶回收,再利用t4连接酶将胶回收产物与ta克隆载体进行连接,连接体系如下:
[0049][0050]
将连接好的产物转化dh5α感受态,制备用于蓝白班筛选的细菌板,涂板隔夜培养后挑选白斑阳性单克隆菌株,每个单克隆细胞至少选取3个白斑,加入含有氨苄西林的细菌培养基以220rpm摇床培养14h后,取菌液送公司测序,获得一株成功敲除细胞株hek293e-apaf1-ko(该细胞株由yko-rp003-hapaf1-1敲除载体获得),测序结果见图4。
[0051]
5.以qpcr、western blot分别鉴定apaf1基因敲除情况
[0052]
分别提取hek293e-wt、hek293e-apaf1-ko株的rna,并进行逆转录反应,获得cdna,具体反应如下:
[0053]
(一)第一步反应体系:
[0054][0055][0056]
(二)将第(一)步所得反应液按下述体系进行:
[0057][0058]
50℃反应15min,85℃5s终止反应
[0059]
设计apaf1基因的qpcr上下游引物,如seq id no.8、seq id no.9所示,对hek293e-wt、hek293e-apaf1-ko株进行qpcr实验检测,将上下游引物稀释10倍,cdna稀释4倍,反应体系如下,结果如图5所示。
[0060][0061]
使用蛋白裂解液(pmsf:ripa=1:100)裂解细胞,收集细胞后进行离心,离心后使用bca法测定蛋白浓度,将20μg蛋白样品加入孔中,上样结束后开始电泳,前30min电压设置为60v,之后电压升高到120v;采用湿转法进行转膜约2h;5%脱脂牛奶封闭2h;孵育一抗,4℃过夜;回收一抗后用tbst洗膜3次,每次10min,室温孵育二抗2h,回收二抗后用tbst洗膜3次,每次10min;使用ecl显色液a液和b液进行显色,结果如图5所示,hek293e-apaf1-ko细胞系apaf1的mrna表达水平、蛋白表达水平均明显低于hek293e-wt细胞,表明apaf1敲除成功。
[0062]
6.hek293e-apaf1-ko的生物特性验证
[0063]
对hek293e-wt、hek293e-apaf1-ko株进行悬浮培养,两株细胞分别按照60万/ml的密度进行悬浮培养,每日进行计数。结果如图6所示,从第3天开始,hek293e-apaf1-ko敲除
细胞系悬浮培养的细胞数量均明显高于hek293e-wt细胞。
[0064]
实施例2apaf1基因敲除hek293细胞系在重组蛋白生产中的应用
[0065]
本实施例apaf1基因敲除hek293细胞系在重组蛋白生产中的应用,通过转染表达相关目的蛋白的质粒(所用重组蛋白表达质粒均由本课题组构建保存,构建方法为本领域常规方法),进行瞬时及稳定表达蛋白的检测,证明hek293e-apaf1-ko细胞可以提高重组蛋白产量,具体实施如下:
[0066]
1、碱性磷酸酶蛋白(seap)瞬时表达
[0067]
选择seap蛋白表达质粒进行细胞转染,分别将hek293e-wt、hek293e-apaf1-ko细胞按50万/ml在6孔板铺板,细胞覆盖率达80%时进行质粒转染,转染剂量2μg/ml,转染6h后进行换液,继续培养48h后进行细胞计数,按60万/ml进行悬浮培养,悬浮培养7日后收样,10000rpm离心10min,收取上清,使用seap检测试剂盒进行检测,加样后在37℃培养箱孵育15min,检测450nm处od值,根据标准曲线计算seap表达量。结果如图7所示,由图中可以看出,与hek293e细胞相比,hek293e-apaf1-ko细胞可以明显提高seap的表达水平。
[0068]
2、玻连蛋白(vn)稳定表达
[0069]
将vn蛋白表达质粒进行细胞转染,检测apaf1基因敲除的hek293e细胞对蛋白稳定表达的影响。分别将hek293e-wt、hek293e-apaf1-ko细胞按照50万/ml的密度在6孔板中铺板,细胞覆盖率达80%时进行质粒转染,转染剂量2μg/ml,转染6h后进行换液,继续培养,转染48h后更换为杀稻瘟菌浓度为7μg/ml培养基进行培养,每隔2-3日更换培养基,筛选3周后得到cell pool,并扩大培养,按照60万/ml的密度进行悬浮培养,悬浮培养7日后收样,10000rpm离心10min,收取上清,取部分上清样品进行蛋白浓缩,进行western blot检测。将20μg蛋白样品加入孔中,上样结束后开始电泳,前30min电压设置为60v,之后电压升高到120v;采用湿转法进行转膜约2h,5%脱脂牛奶封闭2h;室温孵育带有his标签的一抗孵育4℃过夜,回收一抗后用tbst洗膜3次,每次10min;室温孵育鼠二抗2h,回收二抗后用tbst洗膜3次,每次10min;使用ecl显色液a液和b液进行显色。结果如图8所示,由图中可知看出,与hek293e细胞相比,hek293e-apaf1-ko细胞可以明显提高vn的表达水平。
[0070]
最后说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1