一种一体化文库构建的方法及其应用与流程
本发明属于生物,涉及一种一体化文库构建的方法及其应用。
背景技术:
1、从第一代dna测序技术(sanger法)诞生至今,dna测序技术历经数十年的发展和进步,高通量测序技术(high-throughput sequencing technology)又称下一代测序技术(next generation sequencing,ngs),已逐渐成为测序技术的主流并被广泛地应用于生命科学研究的各个领域。尽管随着测序技术的不断更新与普及,测序成本也越来越低,但高深度全基因组测序(30×测序深度)技术本身的花费还是非常昂贵。一个较好的解决方案是将感兴趣的目标区域富集出来后再进行高通量测序。
2、传统的捕获测序技术,通常是将高通量测序文库构建好后,利用探针进行目标区域的文库富集再测序;或者通过多重pcr进行目标区域富集捕获,构建多重扩增子文库后再进行高通量测序。cn111961707a公开了一种基于全基因组酶切捕获的高通量测序技术retseq,利用特殊的内切酶组合,特异性酶切捕获全基因组上的部分区域,实现经济高效的全基因组cnv和snp检测。但上述序列捕获技术均有自身局限性,探针捕获和多重pcr捕获只能针对少数目标基因区域展开,不能均匀覆盖全基因组;retseq技术能均匀覆盖全基因组代表性区域,但对少数特殊单基因病基因区域可能存在位点数量少的问题而造成漏检。
3、胚胎植入前遗传学检测(preimplantation genetic testing,pgt)是指在体外受精的胚胎移植前,取胚胎的遗传物质进行分析,诊断是否有异常,筛选健康胚胎移植,防止遗传病传递的方法。pgt检测主要包括三种类型即染色体非整倍体(pgt-a)、染色体结构异常(pgt-sr)以及单基因病(pgt-m)。目前临床上基于ngs技术的pgt-a、pgt-m、pgt-sr检测分别采用不同的技术手段,尤其是pgt-m、pgt-sr,需要针对不同的变异类型单独设计测试方案,操作流程复杂、周期长,且容易检测失败。retseq技术虽能均匀覆盖全基因组代表性区域,却可能因某些特殊区域位点数量少而造成漏检,这可能是因这些区域位置特殊,如分布在染色体端粒、着丝粒附近,或者存在高度重复序列所致。
4、因此,亟需开发一种一体化文库构建的方法,实现均匀覆盖全基因组cnv和snp,对漏检遗传病区域进行针对性的捕获富集。
技术实现思路
1、针对现有技术的不足和实际需求,本发明提供一种一体化文库构建的方法及其应用,实现了在均匀覆盖全基因组cnv和snp的同时,对漏检遗传病区域进行针对性的捕获富集,提供了一种更完善和准确的一体化胚胎植入前遗传学检测方法。
2、为达到此发明目的,本发明采用以下技术方案:
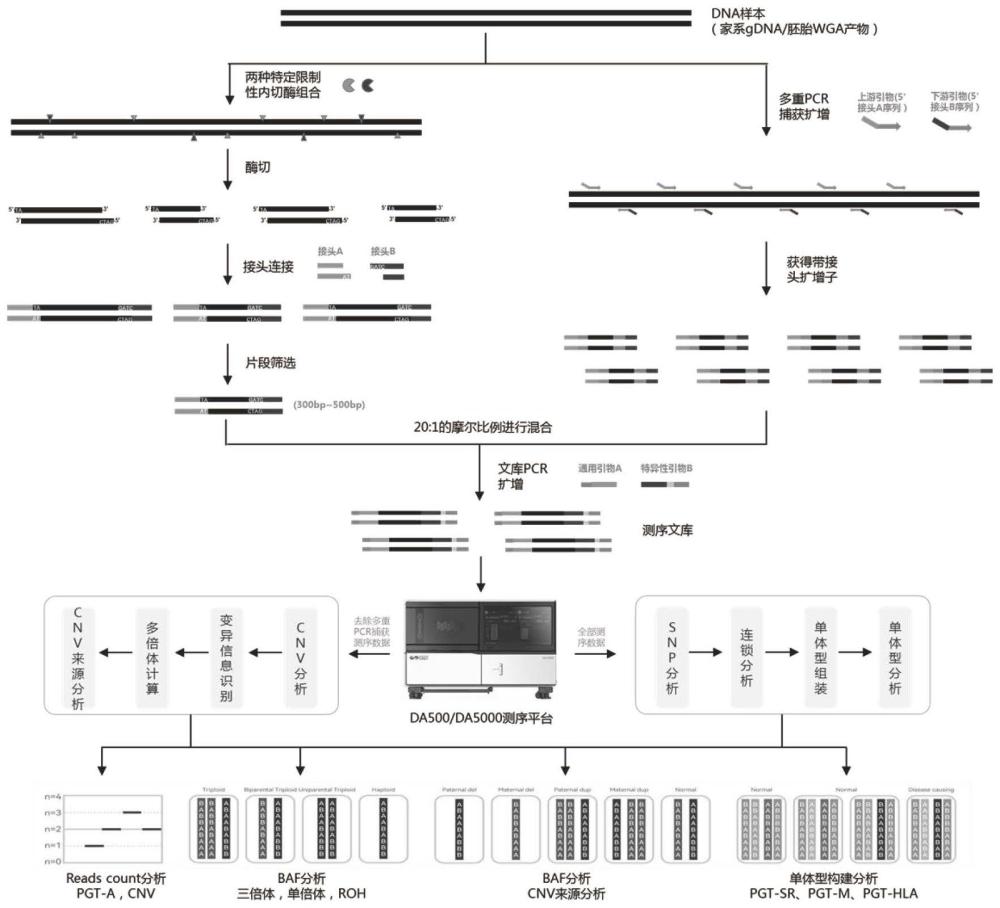
3、第一方面,本发明提供了一种一体化核酸文库构建的方法,所述方法包括:使用第一核酸内切酶和第二核酸内切酶对基因组dna进行酶切得到酶切产物,将酶切产物与测序接头连接,得到连接产物,将筛选后的连接产物与多重pcr扩增得到的扩增子库混合,使用引物进行文库pcr反应,即得所述一体化核酸文库;所述第一核酸内切酶包括bfai;所述第二核酸内切酶包括mboi;所述多重pcr扩增子库由一端含有bfai酶切位点识别序列,另一端含有mboi酶切位点识别序列的dna片段组成。
4、本发明方法实现了在均匀覆盖全基因组cnv和snp的同时,对漏检遗传病区域进行针对性的捕获富集,提供了一种更完善和准确的一体化胚胎植入前遗传学检测方法。
5、优选地,所述多重pcr扩增包括上游引物和下游引物。
6、优选地,所述上游引物的5’端具有和接头a一致的序列。
7、优选地,所述上游引物的5’端包含bfai酶切识别位点ctag序列。
8、优选地,所述下游引物的5’端具有和接头b一致的序列。
9、优选地,所述下游引物的5’端包含mboi酶切识别位点gatc序列。
10、优选地,所述上游引物的核酸序列包括seq id no:1所示的序列;所述下游引物的核酸序列包括seq id no:2所示的序列。
11、seq id no:1:
12、gaacgacatggctacgatccgactt+ctag+位点特异性引物f序列。
13、seq id no:2:
14、ttgtcttcctaagaccgcttggcctccgactt+gatc+位点特异性引物r序列。
15、本发明中,用于多重pcr扩增的上游引物和下游引物针对目的基因致病位点上下游1mb-2 mb范围内的snp位点设计,所述snp位点af值≥0.2,捕获的上游或下游snp位点数均≥30个;所设计多重pcr引物间不存在交叠错配,扩增子长度100bp-300bp,扩增子间最小间距>300bp。所用多重pcr捕获测序技术,可以针对酶切捕获测序技术漏检区域基因设计,酶切捕获测序技术在人基因组上漏检区域不到2%,涉及约50个单基因病相关基因,大多位于端粒、着丝粒或高度重复区域附近。所用多重pcr捕获测序技术,采用基因数量匹配调控机制,根据每次检测需要,挑选对应单基因病相关基因的引物族,混合成引物池,引物池优选1管,最多2管,引物池包含的引物对数量70对-3500对,平均每个基因约70对引物。
16、通过上述引物设计原则,本发明的多重pcr扩增,实现了对bfai和mboi酶切捕获建库流程的兼容性,并通过设计筛选控制引物对上其他区域及扩增子区域的酶切识别位点存在情况,保证一体化建库流程和生信分析流程不受干扰。
17、优选地,所述筛选后的连接产物与扩增子库的摩尔质量比为(10-100):1。
18、上述10-100中的具体点值可以选择10、20、30、40、50、60、70、80、85、90、95、100等。
19、优选地,所述筛选后的连接产物的长度为300bp-500bp。
20、上述300bp-500bp中的具体点值可以选择300bp、320bp、340bp、360bp、380bp、400bp、420bp、460bp、480bp、500bp等。
21、优选地,所述接头a的核酸序列包括seq id no:3和seq id no:4所示的序列。
22、seq id no:3:gaacgacatggctacgatccgacttctag。
23、seq id no:4:aagtcggatcgtagccatgtcgttc。
24、优选地,所述接头b的核酸序列包括seq id no:5和seq id no:6所示的序列。
25、seq id no:5:gatcaagtcggaggccaagcggtcttaggaagacaa。
26、seq id no:6:ttgtcttcctaagaccgcttggcctccgactt。
27、本发明在da500/da5000测序平台的两个接头上分别增加了ctag和gatc的粘性末端,从而可与bfai和mboi酶切产生的粘性末端互补。
28、可以理解,用于改造的原始接头序列包括但不限于适用赛默飞、illumina、华大等测序平台的接头序列,增加的粘性末端序列可根据不同的内切酶进行调整。
29、优选地,所述引物包括通用引物a和特异性引物b;所述通用引物a具有与所述接头a互补配对的序列;所述特异性引物b具有与所述接头b互补配对且具有barcode的序列。
30、在pcr文库扩增的过程中即可带入特定barcode信息,当待测的dna分子来自多个受试样品时,每个样品可以被加上不同的标签序列(barcode),以用于在测序过程中进行样品的区分,从而实现同时对多个样品进行测序。
31、优选地,所述通用引物a的核酸序列包括seq id no:7所示的序列,所述特异性引物b的核酸序列包括seq id no:8所示的序列。
32、seq id no:7:
33、gaacgacatggctacga。
34、seq id no:8:
35、tgtgagccaaggagttg(barcode)ttgtcttcctaagaccgc。
36、第二方面,本发明提供了一种植入前胚胎染色体结构异常分析方法,所述方法包括:采用第一方面所述的一体化核酸文库构建的方法构建胚胎样本和所述胚胎样本的至少一种亲本的测序文库,然后进行测序,根据测序结果分析所述胚胎样本的染色体。
37、优选地,所述结果分析包括:分析染色体结构异常和分析染色体非整倍体异常。
38、本发明中,对一体化测序文库进行ngs上机测序(da500/da5000测序平台),pe100-40m,下机数据进行生物信息学分析,实现对全基因组cnv、单基因病、染色体结构异常、特殊遗传病的一体化检测。
39、可以理解,根据研究需要可调整测序模式和数据量,如可用se100测序、se150测序、pe150测序等模式,数据量也可以10m、20m、30m、40m、50m、60m、70m、80m、100m、120m、160m等。不同数据量下的检测分辨率和有效位点数量会有所不同。
40、本发明中,下机数据进行生物信息学分析时,存在cnv分析和snp分析,snp分析的时候同时使用酶切捕获测序数据和多重pcr捕获测序数据进行分析;cnv分析的时候,先利用reads两端已知引物序列去除多重pcr捕获测序数据(read中任意一端存在已知引物序列即可),再使用酶切捕获部分的测序数据进行cnv分析。
41、与现有技术相比,本发明具有如下有益效果:
42、(1)本发明特异筛选的限制性内切酶组合包括bfai和mboi,酶切识别位点分别为“ctag”和“gatc”,这两个酶的核心识别序列包括了agct四种碱基,且每种碱基的占比均相等,通过选择ctag和gatc两种不同的碱基排序组合,能够保证全基因组的均匀覆盖和酶切片段的合适分布,所用bfai和mboi在人基因组上酶切捕获的snp数量>80万个,覆盖>98%的窗口,且分布均匀无明显偏好,酶切捕获效果稳定;
43、(2)本发明能够实现对bfai和mboi酶切捕获建库流程的兼容性,保证了一体化建库流程和生信分析流程不受干扰;
44、(3)本发明将多重pcr扩增子库与酶切分选片段按照特定比例混合,再一同pcr扩增获得一体化文库,使用同一生信分析流程进行数据分析,将两种独立的检测流程合二为一,极大节约了成本和时间。
- 还没有人留言评论。精彩留言会获得点赞!