一种GalNAc化合物、其与寡核苷酸缀合物及制备方法与流程
本发明属于化学合成,涉及一种galnac亚磷酰胺化合物、其合成及其与寡核苷酸缀合方法。
背景技术:
1、小核酸药物(寡核苷酸)凭借其自身独特的性质正逐步发展成为传统小分子药物的替代物,用以调控与疾病相关蛋白的功能。寡核苷酸可用于沉默或激活特定疾病的基因表达,从而防止或促进特定蛋白的形成,起到治疗疾病的作用。寡核苷酸包括但不限于反义寡核苷酸(aso),小干扰rna(sirna)、小激活rna(sarna)和微小rna(microrna,mirna)。由于寡核苷酸药物疗效较好,技术取得突破,成为当前最受关注的一类技术,目前全球已有数款药物获批上市。
2、尽管sirna分子具有较好的治疗效用,但sirna在体内的递送仍然是一个重大挑战,因为它会被核酸酶快速降解、细胞摄取不良以及全身给药后肾脏清除速度很快。因此需要运载工具将sirna分子运输到靶组织细胞中的作用位点,开发一种安全可靠的方法来选择性地靶向患病器官和组织仍然是将其转化为临床的关键需求。早期方法侧重于脂质纳米粒子(lnp)和合成纳米粒子,以解决sirna递送问题,而n-乙酰半乳糖胺(galnac)和sirna缀合物的递送平台因其针对肝靶向性肝素递送的效力和安全性受到广泛的关注和研究。研究发现去唾液酸糖蛋白受体(asgpr)是肝细胞表面特异性表达的一种内吞性受体,主要存在于肝脏实质细胞朝向窦状隙一侧的细胞膜表面,可以特异性的结合糖类。半乳糖(gal)和n-乙酰半乳糖胺(n-acetylgalgactosamine,galnac),是一种与肝表面去唾液酸糖蛋白受体(asgpr)结合的配体。其中,galnac与asgpr结合的亲和性比gal高约50倍。研究发现,其亲和力的次序依次为:四触角>三触角>>双触角 >>单触角半乳糖苷。近年来,利用asgpr的高亲和性配体n-乙酰半乳糖胺(galnac)作为靶向分子,在核酸药物的肝靶向递送方面取得了一定的进展。多种galnac可以实现与sirna和aso的缀合,利用这种技术已经开展了淀粉样病变、血友病、高胆固醇血症、乙型肝炎等疾病的药物开发。
3、综上所述,galnac与寡核苷酸的缀合取得了一定的进展,主要包括galnac亚磷酰胺方法和后期修饰的方法,但是目前的缀合对反应的基团存在一定的限制(比如空间位阻效应,序列位点等),因此相对比较局限。
技术实现思路
1、针对现有技术的不足和实际需求,本发明提供了一种galnac化合物、其与寡核苷酸缀合物及制备方法,设计特定galnac化合物,可实现与寡核苷酸任意位置进行缀合,推动小核酸药物的应用。
2、为达此目的,本发明采用以下技术方案:
3、第一方面,本发明提供一种galnac化合物,所述galnac化合物包括式i所示的化合物或其化学上可接受的盐;
4、式i
5、其中,l1为-hnc(o)-或-c(o)nh-;l2为-(ch2)n1-,其中n1为1-7的整数;z1为o或c;l3为-(ch2)n2-,其中n2为1-4的整数;z2为o或c;n为0-10的整数;l4为-(ch2)n3-,其中n3为1-7的整数;a的结构式为式ii或式iii,其中b为核苷酸的碱基部分,包括天然存在的核碱基和核碱基类似物;r1为亚磷酰胺化合物或氢;r2为羟基保护基团;r3为氢、烷氧基或卤素及其类似物;
6、式ii 式iii
7、 g的结构式为式iv;
8、式iv
9、其中,x1为-(ch2)a-或-(ch2ch2o)ach2-,a为1-5的整数;x2为-(ch2)b-,b为1-6的整数;y1为0或1;y2为0、1或2;y3为1、2或3。
10、本发明中,设计新型的galnac化合物,可实现与寡聚核酸任意位置连接,且够结合脱唾液酸糖蛋白受体(asgpr),可有效应用于寡聚核酸的递送,增强递送效果,提高寡聚核酸作用效果以及在体内的半衰期、药峰浓度和药-时曲线下面积等,推动寡聚核酸药物的发展。
11、在一种实施方案中,l1为-hnc(o)-。
12、在一种实施方案中,l2为-(ch2)4-。
13、在一种实施方案中,z1为c。
14、在一种实施方案中,l3为-(ch2)2-。
15、在一种实施方案中,z2为c。
16、在一种实施方案中,n为1。
17、在一种实施方案中,l4为-(ch2)2-。
18、在一种实施方案中,所述亚磷酰胺化合物的结构式为式v。
19、式v。
20、可选地,所述r2为4, 4'-二甲氧基三苯甲基、单甲氧基三苯甲基、三苯甲基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、三异丙基甲硅烷基或异丙基二甲基甲硅烷基中任意一种。
21、可选地,所述a的结构式为式vi或式vii。
22、式vi 式vii。
23、可选地,所述g的结构式为式viii。
24、式viii。
25、在一实施方案中,所述galnac化合物的结构式如式ix(命名为yk-gal-101)、式x(命名为yk-gal-102)或式xi(命名为yk-gal-103)所示。
26、式ix
27、式x
28、式xi。
29、第二方面,本发明提供一种缀合物,所述缀合物由寡核苷酸和第一方面所述的galnac化合物缀合得到,结构式包括式xii所示结构。
30、式xii
31、式xii中,oligo代表寡核苷酸,其余部分代表galnac化合物部分,所述galnac化合物部分中的g、l1、l2、z1、l3、z2、n和l4同权利要求1,所述galnac化合物部分与所述寡核苷酸的5’端、中间位置或3’端缀合。
32、可选地,所述寡核苷酸包括非硫代寡核苷酸和硫代寡核苷酸。
33、可选地,所述寡核苷酸包括小干扰核苷酸、dna、微小rna、小激活rna、小向导rna、转运rna、反义核苷酸或适配体中任意一种或至少两种的组合。
34、可选地,所述寡核苷酸调节靶基因的表达。
35、可选地,所述寡核苷酸和所述galnac化合物部分通过键或可切割的连接体连接。
36、本发明中,利用设计的galnac化合物与寡聚核酸任意位置缀合,可构建递送平台,实现良好地递送,可以理解,所述中间位置指寡聚核酸链上除5’端和3’端外的中间的任意位置。
37、第三方面,本发明提供第二方面所述缀合物的制备方法,所述方法包括:
38、将第一方面所述的galnac化合物与固相支持物相连,得到偶联物;利用所述偶联物为固相载体,采用化学固相合成法合成寡核苷酸,得到所述缀合物。
39、本发明中,基于设计的新型galnac化合物,开发化学固相合成方法进行缀合,可实现快速、高效制备缀合物。
40、可以理解,化学固相合成法包括粗品合成、脱保护、纯化等步骤。
41、可选地,在粗品合成步骤中,涉及galnac化合物的偶合时间为15~25 min,包括但不限于16、17、18、19、20、21、22、23或24 min等等。
42、可选地,在粗品合成步骤中,涉及galnac化合物的抽液配制循环3~8次。
43、可选地,在粗品合成步骤中,涉及galnac化合物的抽液配制循环4-5次。
44、可选地,在粗品合成步骤中,涉及galnac化合物的使用量相比固相支持物的载量为15-30倍摩尔比例,包括但不限于16、17、18、19、20、21、22、25、26、28或29倍摩尔比例等等。
45、可选地,在粗品合成步骤中,涉及galnac化合物的使用量相比固相支持物的载量为18-25倍摩尔比例。
46、可选地,所述寡核苷酸包括非硫代寡核苷酸和硫代寡核苷酸。
47、可选地,要制备galnac化合物与所述寡核苷酸的3’端缀合物,所述galnac化合物中r1为h;要制备galnac化合物与所述寡核苷酸的5’端缀合物,所述galnac化合物中a的结构式为式iii;要制备galnac化合物与所述寡核苷酸的中间位置缀合物,所述galnac化合物中a的结构式为式ii。
48、第四方面,本发明提供第二方面所述的缀合物在制备药物中的应用。
49、本发明中,利用设计的galnac化合物与寡聚核酸任意位置缀合,可构建递送平台,实现良好地递送,进而可有效应用于寡聚核酸药物等的开发,包括降血脂药物,治疗乙肝及高血压的药物等。
50、第五方面,本发明提供一种药物组合物,所述药物组合物包括第二方面所述的缀合物以及药学上可接受的辅料。
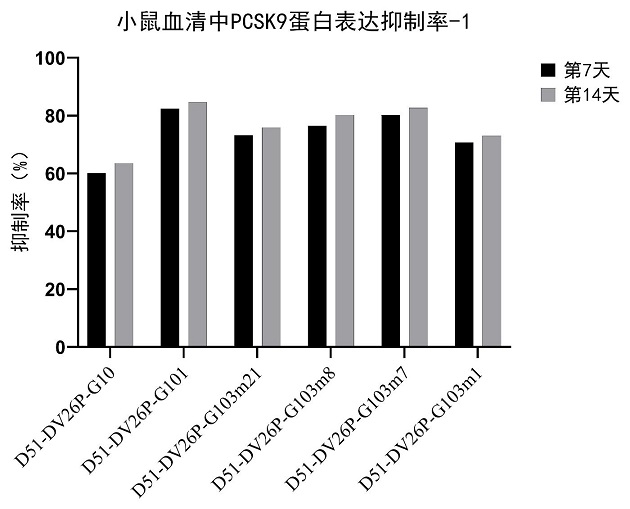
51、可选地,所述辅料包括载体、防腐剂、抑菌剂或抗氧化剂等。
52、与现有技术相比,本发明具有以下有益效果:
53、1.本发明设计新型的galnac化合物,可与寡核苷酸序列的任意位置连接,而现有技术(例如将galnac配体修饰到寡核苷酸3’端上(us20150119444a1, us20150119445a1);通过液相合成方法将galnac配体连接到寡核苷酸5’端上(us20150126718a1))只是将galnac化合物连接到寡核苷酸两端(3’或5’端);
54、本发明将galnac化合物与核糖环2’端进行连接,然后合成galnac亚磷酰胺单体化合物,再利用固相方法合成galnac修饰的寡核苷酸,此方法可以将一个或多个galnac化合物修饰到寡核苷酸序列的任意位置;
55、2.本发明能够通过固相合成方法,实现galnac化合物与寡核苷酸序列的任意位置连接,如制备得到5’端galnac修饰的寡核苷酸,相比于现有液相方法(如us20150126718a1),减少了反应步骤,缩短了反应时间,并且实验后处理更加简单,收率可以达到70%以上,纯度可以达到98%以上;
56、3. 由本发明galnac化合物缀合到sirna的5’端或中间任意位点的galnac-sirna缀合物,与现有技术3’端galnac缀合的sirna相比,对肝组织递送效果、靶标蛋白表达的抑制率、靶标mrna的抑制率和ldl-c的降低水平,以及对药物肝脏中的半衰期、药峰浓度和药-时曲线下面积等指标均有显著提升;
57、例如本发明具体实施例中,与d51-dv26p-g10相比,d51-dv26p-g101对小鼠血清中pcsk9蛋白抑制率提高了22.3%,对小鼠肝脏中pcsk9 mrna抑制率提高了23.4%,小鼠血清中的ldl-c降低水平提高了14.2%,肝脏中半衰期增长了26.03%,药峰浓度提高了14.05%,药-时曲线下面积增加了20.26%;
58、与d5-dv26p-g302相比,d5-dv26p-g103m8对小鼠血清中pcsk9蛋白抑制率提高了27.0%,对小鼠肝脏中pcsk9 mrna抑制率提高了23.4%,小鼠血清中ldl-c降低水平提高了17.2%,肝脏中半衰期增长了21.43%,药峰浓度提高了8.82%,药-时曲线下面积增加了14.27%;
59、4.与现有技术galnac配体化学结构不同,本发明将galnac配体制备成亚磷酰胺单体,然后通过固相合成方法将修饰的galnac配体直接连接到寡核苷酸序列上,与现有技术(如us20150119444a1, us20150119445a1、us20150126718a1等)相比,操作更简单、高效。
- 还没有人留言评论。精彩留言会获得点赞!