能够即时可视化检测次氯酸的碳点及其制备方法和应用与流程
本发明涉及碳点,具体地,涉及一种能够即时可视化检测次氯酸的碳点及其制备方法和应用。
背景技术:
目前,检测次氯酸的方法有电化学法和荧光法等。其中,荧光方法由于其在操作简便、非侵入性、高的时间和空间分辨能力等优点,应用于各类检测体系。多种荧光探针已被用于次氯酸的检测,主要包括有机小分子、半导体量子点和纳米贵金属团簇等。
然而,这些探针存在着水溶性差、光漂白性和对环境具有潜在危害等缺点。相反,碳点具有良好的水溶性、光稳定性和低毒性,能够应用于各种领域。当前,用于检测次氯酸的碳点均是基于“turn-off”模式,反应前后无颜色变化,难以实现快速地可视化检测。
技术实现要素:
本发明的目的是提供一种能够即时可视化检测次氯酸的碳点及其制备方法和应用,该碳点对次氯酸有高选择性、高灵敏度和即时的可视化响应,进而使其能够应用于次氯酸可视化检测,该制备方法操作简单、成本低廉、高的可操作性和重复性的优点。
为了实现上述目的,本发明提供了一种能够即时可视化检测次氯酸的碳点的制备方法,包括:
1)将间氨基苯酚水溶液去除溶解氧后,在惰性气体的保护下进行水热反应以得到棕褐色液体;
2)将棕褐色液体进行透析,接着将透析袋内的溶液浓缩得到棕黄色液体,然后将棕黄色液体经过色谱柱层析处理得到碳点胶体溶液。
本发明还提供了一种能够即时可视化检测次氯酸的碳点,该碳点通过上述的制备方法制备而得。
本发明进一步提供了一种如上述的碳点在可视化检测次氯酸中的应用。
在上述技术方案中,本发明以间氨基苯酚为前驱体,选择优化的水热合成方法及分离方案,获得了组分单一、具备高亮度和光稳定性的碳点。
该碳点具有如下优点:(1)碳点表面具有多个酚羟基识别位点,该识别位点能与次氯酸反应,发生从酚到酮的结构变化,导致碳点的电子态的变化,进而荧光发射从黄绿色变为蓝色;(2)反应前后荧光发射发生显著的颜色变化(从黄绿到蓝色,107nm蓝移),实现了荧光比率传感;(3)氧化前后的碳点都具有高的荧光量子产率,使得荧光颜色变化易于观察;(4)该荧光比率传感响应速度快,能够在1秒内完成反应,可实现即时检测。
该碳点的制备方法操作简单、成本低廉;具有高的可操作性和重复性;分离方法简单高效,分离后组分单一;获得的碳点能够实现对次氯酸的高选择性、高灵敏度和即时的响应,并导致荧光发射颜色的显著变化;进而使碳点能够应用于检测次氯酸。
本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
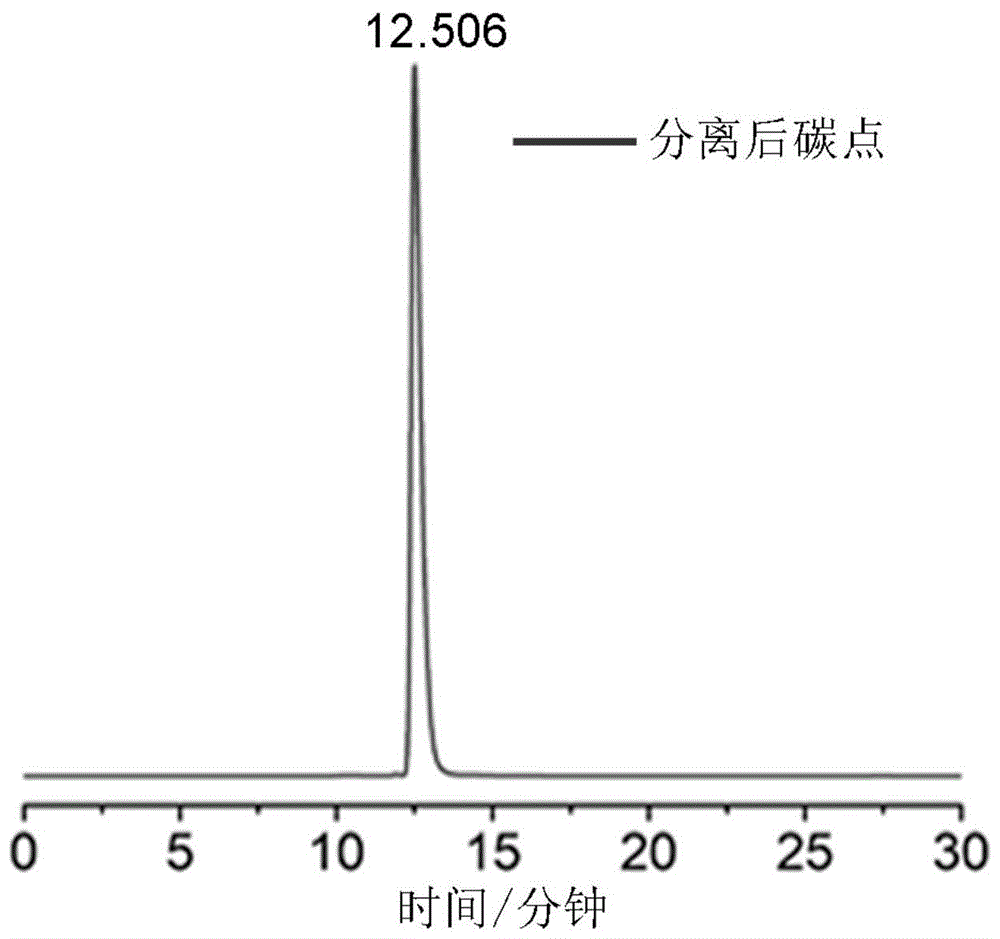
图1是检测例1中高效液相色谱表征图;
图2是检测例1中透射电子显微镜表征图;
图3是检测例1中原子力显微镜表征图;
图4是检测例1中紫外-可见光吸收、荧光激发和荧光发射光谱表征图;
图5是检测例1中对其它种类活性氧的选择性检测图;
图6是检测例1中对次氯酸检测的荧光光谱变化图;
图7是检测例1中对次氯酸荧光检测的标准曲线图;
图8是检测例1中不同浓度次氯酸(5μm、10μm、20μm)荧光检测动力学图。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供了一种能够即时可视化检测次氯酸的碳点的制备方法,包括:
1)将间氨基苯酚水溶液去除溶解氧后,在惰性气体的保护下进行水热反应以得到棕褐色液体;
2)将棕褐色液体进行透析,接着将透析袋内的溶液浓缩得到棕黄色液体,然后将棕黄色液体经过色谱柱层析处理得到碳点胶体溶液。
在本发明中,各物料的用量可以在宽的范围内选择,但是为了考虑原料在水中的溶解度和反应容器的体积,过量的原料将带来剩余及更多的中间体,给分离带来困难。因此,在综合产率和分离效果前提下,优选地,在步骤1)中,间氨基苯酚、水的用量比为0.1635g:40-60ml。
在本发明中,水热反应的条件可以在宽的范围内选择,但是为了目标产物的产率和保留前驱体的结构,较低的温度不能使前驱体充分碳化,较高温度可能破坏表面结构,优选地,在步骤1)中,水热反应满足以下条件:反应温度为130-180℃,和/或,反应时间为8-12h。
在本发明中,去除溶解氧的方式可以采用多种方式,但是为了便于操作同时为了提高溶解氧的去除效果,优选地,在步骤1)中,去除溶解氧是通过向间氨基苯酚水溶液中通入惰性气体20-40min。
在本发明中,为了进一步提高溶解氧的去除效果,优选地,在水热反应之前,将间氨基苯酚水溶液转入反应容器中并通入惰性气体4-10min。
在本发明中,惰性气体的具体种类可以在宽的范围内选择,但是从成本上考虑,优选地,惰性气体选自氮气、氦气、氩气中的至少一者。
在本发明中,透析的条件可以在宽的范围内选择,但是为了进一步提高碳点的纯度,优选地,透析满足以下条件:透析袋的截留分子量为500-1000da,和/或,透析时间为24-48h。
在本发明中,旋转蒸发浓缩的条件可以在宽的范围内选择,但是为了进一步提高碳点的纯度,优选地,透析袋内的溶液是通过旋转蒸发浓缩进行的,其中,旋转蒸发浓缩满足以下条件:浓缩温度为55-70℃。
在本发明中,色谱柱层析处理的条件可以在宽的范围内选择,但是为了进一步提高碳点的纯度,优选地,色谱柱层析处理采用的流动相为体积比为(3-5):1的乙酸乙酯和石油醚的混合溶液。
在本发明中,为了进一步便于保存碳点,优选地,在色谱柱层析处理之后,该制备方法还包括:将碳点胶体溶液旋蒸浓缩、冷冻干燥;
在上述实施方式中,旋转浓缩可以在宽的范围内选择,但是为了去除溶剂的效果,优选地,旋转浓缩满足以下条件:浓缩温度为55-70℃;
在上述实施方式中,旋转浓缩可以在宽的范围内选择,但是为了提高干燥效果,优选地,冷冻干燥满足以下条件:干燥温度为-30~-50℃,干燥压力为10-14pa,干燥时间为1-2天。
本发明还提供了一种能够即时可视化检测次氯酸的碳点,该碳点通过上述的制备方法制备而得。
本发明进一步提供了一种如上述的碳点在检测次氯酸中的应用。
以下将通过实施例对本发明进行详细描述。
实施例1
将0.1635g间氨基苯酚(m-aminophenol,m-ap)溶于50ml超纯水中,通氮气30min除去溶解氧;将m-ap水溶液转入三个25ml反应釜中,再分别通气5min;将反应釜置入鼓风式干燥箱中,在140℃下加热10h;自然冷却后,获得棕褐色液体。
上述棕褐色液体经透析处理(截留分子量为500d)24h后,将袋内的液体在真空条件下旋转蒸发浓缩(浓缩温度为55℃,浓缩时间为1h,转速为70rpm),获得5ml左右棕黄色液体;将棕黄色液体经过色谱柱层析法分离(流动相为体积比为3:1的乙酸乙酯和石油醚的混合液,固定相为硅胶粉(200目)),;将停留在色谱柱内上层黄色组分取出,并用纯水洗脱,获得在紫外灯下发射黄绿色荧光的液体,即为目标产物碳点胶体溶液;碳点溶液经过旋蒸浓缩(浓缩温度为60℃,浓缩时间为30min,转速为70rpm)、冷冻干燥(干燥温度为-40℃,干燥压力为13pa,干燥时间为1-2天)后得到碳点。
实施例2
按照实施例1的方法进行,所不同的是将50ml超纯水换为40ml超纯水。
实施例3
按照实施例1的方法进行,所不同的是将50ml超纯水换为60ml超纯水。
实施例4
按照实施例1的方法进行,所不同的是,将“在140℃下加热10h”更改为“在130℃下加热12h”。
实施例5
按照实施例1的方法进行,所不同的是,将“在140℃下加热10h”更改为“在180℃下加热8h”。
检测例1
1)通过高效液相色谱(安捷伦1200系统)对实施例1的碳点进行高效液相色谱检测,结果见图1,图1中只有一个尖峰,说明其组分单一。
2)通过日立ht-7800显微镜对实施例1的碳点进行透射电子显微镜检测,结果见图2,由图可知,碳点的直径尺寸分布在1.5-4.5nm范围内,平均直径为2.76nm。
3)通过布鲁克dimension3100v显微镜对实施例1的碳点进行原子力显微镜表征,结果见图3,由图可知,碳点的厚度约为2.84nm,说明它们具有球形或准球形结构。
4)通过日立u-3100光谱仪和f-4600光谱仪分别对实施例1的碳点进行紫外-可见光吸收光谱(abs)、荧光激发(ex)和发射光谱表征,结果见图4,由图可知,碳点除了在240–275和325nm处具有典型的c=cπ-π*和c-n/c-on-π*跃迁吸收峰外,在496nm处还有一个显著的特征吸收峰,在自然光下呈现亮黄色;在不同波长的激发光下,其发射峰都处于537nm左右(紫外灯下为黄绿色荧光),表现出不随激发依赖现象。经归一化处理后,在最佳激发下(490nm)的发射光谱与激发光谱呈现镜像对称。此外,该碳点具有非常高的荧光量子产率,通过稳态荧光光谱仪(fls920系统)测出其绝对产率达到74.7%。
5)选择性检测:在5μg/ml的碳点溶液中加入20μm(μm为μmol/l)的常见的活性氧(包括次氯酸、过氧亚硝酸根、羟基自由基、超氧阴离子、单线态氧、过氧化氢、亚硝酸根、一氧化氮),反应10分钟后在360nm激发下测试荧光发射光谱。检测结果见图5,纵坐标为溶液发射光谱在430nm和537nm处荧光强度比值(f430/f537),由图可知,相对于次氯酸,碳点对其他活性氧的响应非常小,说明该碳点对次氯酸的检测具有高选择性。
6)在5μg/ml的碳点溶液中加入不同浓度(0-20μm)的次氯酸,10min后进行荧光检测(360nm波长激发),结果见图6,由图可知,随着次氯酸的加入,碳点的发射光谱在537nm处(黄绿色)的强度降低很小,而在430nm处(蓝色)出现新的荧光发射峰,且其荧光强度显著增强。以浓度为横坐标,f430/f537为纵坐标,其变化规律如图7所示,根据其直线部分(0-5μm)的斜率,可以计算出检测限为8.6nm,说明碳点对次氯酸的检测具有高的灵敏性。
7)在5μg/ml的碳点溶液中,分别加入5μm、10μm和20μm的次氯酸,360nm波长激发下检测荧光发射光谱随时间的变化规律,并以时间(min)为横坐标,f430/f537为纵坐标,分析碳点对次氯酸响应的动力学。结果如图8所示,三种浓度下,f430/f537均在10min完成反应,且在加入5μm次氯酸时,0.5min后反应即进入平台,说明碳点与次氯酸的反应是扩散控制且几乎是瞬时完成的。
按照上述1)-7)的方法对实施例2-5进行相同的检测,检测结果与实施例1的产物的检测结果基本一致。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!