一种具有催化光降解有机染料功能的铕配合物及其制备方法与应用与流程
本发明属于配合物催化材料技术领域,具体涉及一种具有催化光降解有机染料功能的铕配合物及其制备方法与应用。
背景技术:
水体污染是染料生产和使用产业所面临的巨大的环境挑战。考虑到有机染料的使用性能,多数有机染料结构稳定,因此,常规的生物处理染料废水效果不佳。为了有效从废水中去除染料,包括离子交换、凝聚/絮凝、吸附、化学氧化、光催化等物理化学技术引起了环保技术人员的关注。
其中,光催化技术基于原位生成具有高活性的·oh和o2-,将染料分解为co2和h2o,具有反应条件为何、成本低、效率高的优点。作为催化光降解有机染料的关键因素,寻找优良的光催化剂成为了材料研究人员的关注重点。
与传统半导体光催化剂相比,配合物催化剂可以通过改变有机配体和中心金属离子实现其光学性能的调控,且配合物的结构稳定性较高,不会造成使用过程中的二次污染。
目前,一些稀土金属配合物已经用于有机染料的光降解催化,但针对不同稀土金属的、具有多样性结构的铕配合物的相关研究还存在较多空白。
因此,开发一种具有催化光降解有机染料功能的铕配合物成为了本领域技术人员亟需解决的问题。
技术实现要素:
有鉴于此,本发明的目的是针对现有技术中存在的问题,提供一种具有催化光降解有机染料功能的铕配合物。
为了实现上述目的,本发明采用如下技术方案:
一种具有催化光降解有机染料功能的铕配合物,所述铕配合物为c52h44eu2n16o19,分子结构如下所示:
进一步的,所述铕配合物属于属于monoclinic晶系,c2/c空间群,晶胞参数为:
值得说明的是,所述具有催化光降解有机染料功能的铕配合物含有1个eu(ⅲ)离子、4个桥氧的结构,并与h2o中的o原子进行配位,呈现九配位结构,且具有多配位点的多样性结构。
本发明的第二个目的在于,提供所述的具有催化光降解有机染料功能的铕配合物的制备方法。
为了实现上述目的,本发明提供如下技术方案:
所述的具有催化光降解有机染料功能的铕配合物的制备方法,步骤包括:
i、称取na2moo6·2h2o溶解于水中,加入hcl调节体系ph值,搅拌得到溶液a,称取四丁基溴化铵溶于水中,得到溶液b;
ii、向溶液a中加入溶液b,搅拌并加热至80℃,反应45min,抽滤得到黄色沉淀,水洗即得到[(n-c4h9)4n]2[mo6o19]的粗产物;
iii、将[(n-c4h9)4n]2[mo6o19]的粗产物溶解于丙酮中,加热至60℃,冷藏处理24h,收集黄色沉淀抽滤,乙醚洗涤、干燥得到精制[(n-c4h9)4n]2[mo6o19];
iv、称取n-羟乙基-3,3-二甲基-6-硝基吲哚啉螺吡喃、eu(no3)3·6h2o和步骤iii制备得到的[(n-c4h9)4n]2[mo6o19]于烧杯中,依次加入n,n’-二甲基甲酰胺、异丙醇,搅拌,滴加乙二胺溶液调节体系ph=8,于密闭烘箱80℃反应3天,待反应结束后自然冷却至室温,获得的橙黄色长条状晶体即为所述具有催化光降解有机染料功能的铕配合物c52h44eu2n16o19。
值得说明的是,本发明通过水热合成法,以n-羟乙基-3,3-二甲基-6-硝基吲哚啉螺吡喃、镧系稀土金属盐及lindqvsit型[(n-c4h9)4n]2[mo6o19]多金属氧酸盐为原料,合成得到具有催化光降解有机染料功能的铕配合物c52h44eu2n16o19。与预期的过渡金属阳离子直接与螺吡喃分子率先进行配位不同,表征发现,本发明反应体系中的螺吡喃配体首先与乙二胺发生了原位反应,形成席夫碱配体,然后金属eu再与席夫碱配体配位形成九配位的配合物c52h44eu2n16o19。
并且,本发明制备并使用的lindqvsit型多金属氧酸盐[(n-c4h9)4n]2[mo6o19],结构式为[m6o19]n-(其中m=mo),其结构是由6个{mo8}八面体以公边形式相连,且具有oh对称性。
进一步地,所述步骤i中na2moo6·2h2o与四丁基溴化铵的摩尔比为1:1.875,且溶液a的浓度为10mmol/l,溶液b的浓度为18.75mmol/l。
进一步地,所述步骤ii中溶液a与溶液b的体积比为5:1。
进一步地,所述步骤iv中n-羟乙基-3,3-二甲基-6-硝基吲哚啉螺吡喃、eu(no3)3·6h2o和[(n-c4h9)4n]2[mo6o19]的摩尔比为1:30:10,所述n,n’-二甲基甲酰胺与异丙醇的体积比为1:3,且所述[(n-c4h9)4n]2[mo6o19]的浓度为0.008mol/l。
本发明的第三个目的在于提供所述的具有催化光降解有机染料功能的铕配合物的应用。
为了实现上述目的,本发明提供如下技术方案:
所述铕配合物在催化光降解有机染料中的应用。
进一步地,所述有机染料包括盐酸副玫瑰苯胺、亚甲基蓝、罗丹明b或甲基橙。
本发明与现有技术相比,具有以下优点和有益效果:
1、本发明独创性地合成了一种金属eu与席夫碱配体配位形成九配位的具有催化光降解有机染料功能的铕配合物c52h44eu2n16o19,该铕配合物打破了螺吡喃类配体会直接与过渡金属阳离子发生配位反应的认知,在螺吡喃类配体、乙二胺和过渡金属阳离子共混的情况下,首先由螺吡喃配体与乙二胺发生原位反应形成席夫碱配体,然后再与金属eu形成九配位化合物。
2、本发明提供的铕配合物c52h44eu2n16o19,光催化降解性能优异且结构稳定,可以在紫外光照射下实现盐酸副玫瑰苯胺、亚甲基蓝、罗丹明b或甲基橙的光催化降解。
3、本发明提供的水热合成法制备方法简单、原料易得、合成步骤少、合成条件温和,产率较高,具有产业化应用的潜力。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图
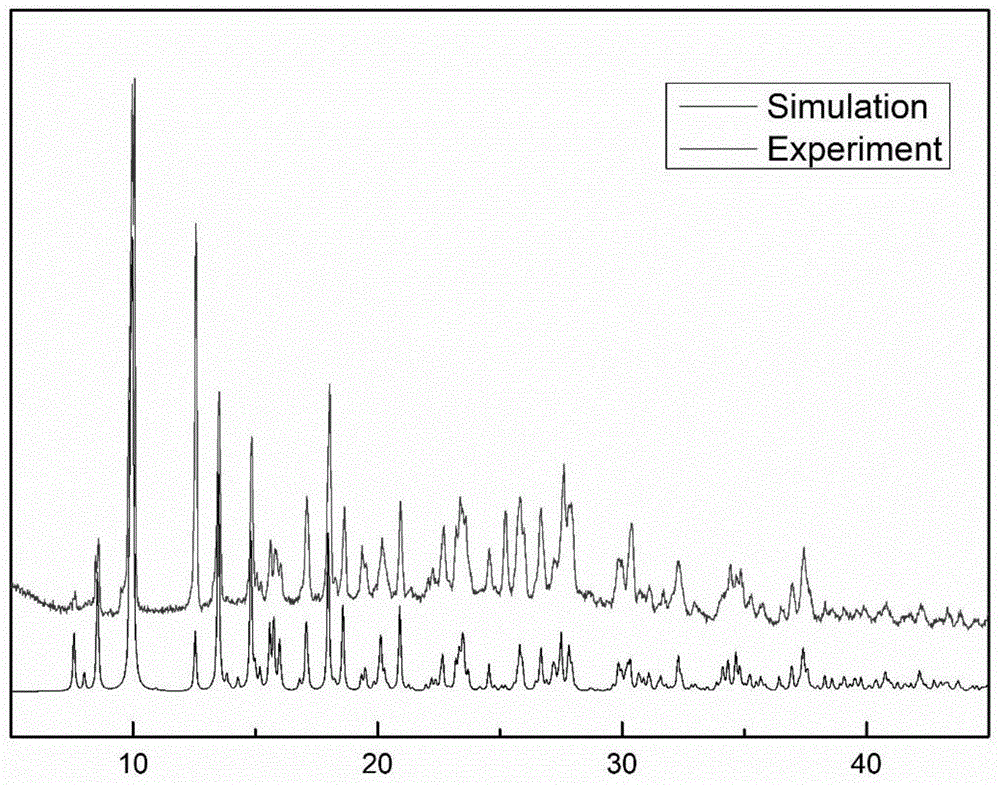
图1为本发明实验例1测定的具有催化光降解有机染料功能的铕配合物粉末x-射线衍射图:理论和实验的对比图。
图2为本发明实验例3测定的具有催化光降解有机染料功能的铕配合物的ir光谱图。
图3为本发明实验例4测定的具有催化光降解有机染料功能的铕配合物的的紫外光谱图。
图4为本发明实验例5测定的具有催化光降解有机染料功能的铕配合物的荧光图。
图5为本发明实验例5测定的具有催化光降解有机染料功能的铕配合物的能级匹配分析图。
图6为本发明实施例2提供的具有催化光降解有机染料功能的铕配合物(2mg)在亚甲基蓝的光降解图。
图7为本发明实施例2提供的具有催化光降解有机染料功能的铕配合物(5mg)对亚甲基蓝的光催化降解的紫外吸收光谱图。
图8为本发明实施例2提供的具有催化光降解有机染料功能的铕配合物(8mg)对亚甲基蓝的光催化降解的紫外吸收光谱图。
图9为本发明实施例2提供的具有催化光降解有机染料功能的铕配合物对亚甲基蓝的光降解趋势图。
图10为本发明实施例3提供的具有催化光降解有机染料功能的铕配合物(2mg)在甲基橙的光降解图。
图11为本发明实施例3提供的具有催化光降解有机染料功能的铕配合物(5mg)对甲基橙的光催化降解的紫外吸收光谱图。
图12为本发明实施例3提供的具有催化光降解有机染料功能的铕配合物(8mg)对甲基橙的光催化降解的紫外吸收光谱图。
图13为本发明实施例3提供的具有催化光降解有机染料功能的铕配合物对甲基橙的光降解趋势图。
图14为本发明实施例4提供的具有催化光降解有机染料功能的铕配合物(2mg)在盐酸副玫瑰苯胺的光降解图。
图15为本发明实施例4提供的具有催化光降解有机染料功能的铕配合物(5mg)对盐酸副玫瑰苯胺的光催化降解的紫外吸收光谱图。
图16为本发明实施例4提供的具有催化光降解有机染料功能的铕配合物(8mg)对盐酸副玫瑰苯胺的光催化降解的紫外吸收光谱图。
图17为本发明实施例4提供的具有催化光降解有机染料功能的铕配合物对盐酸副玫瑰苯胺的光降解趋势图。
图18为本发明实施例5提供的具有催化光降解有机染料功能的铕配合物(2mg)在罗丹明b的光降解图。
图19为本发明实施例5提供的具有催化光降解有机染料功能的铕配合物(5mg)对罗丹明b的光催化降解的紫外吸收光谱图。
图20为本发明实施例5提供的具有催化光降解有机染料功能的铕配合物(8mg)对罗丹明b的光催化降解的紫外吸收光谱图。
图21为本发明实施例5提供的具有催化光降解有机染料功能的铕配合物对罗丹明b的光降解趋势图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为更好地理解本发明,下面通过以下实施例对本发明作进一步具体的阐述,但不可理解为对本发明的限定,对于本领域的技术人员根据上述发明内容所作的一些非本质的改进与调整,也视为落在本发明的保护范围内。
实施例1
一种具有催化光降解有机染料功能的铕配合物及其制备方法,称取na2moo6·2h2o2.5g溶解于10ml水,加入6mhcl3ml溶液调节体系的酸度,搅拌得溶液a。称取四丁基溴化铵1.2g溶解于2ml水中,为溶液b。向溶液a加入溶液b后,搅拌,得白色浑浊液。将含白色浑浊液加热至80℃,搅拌至转变为黄色沉淀。抽滤后得黄色沉淀,用20ml水洗洗涤黄色沉淀,即为粗产物。将粗产物溶解于80ml丙酮中,加热至60℃,搅拌直至完全溶解,即得溶液c。溶解后冷却至室温时,将溶液c进行冷藏处理达24h,收集黄色沉淀。
抽滤后可得黄色固体,用20ml乙醚洗涤黄色固体,干燥,为lindqvsit型多金属氧酸盐:[(n-c4h9)4n]2[mo6o19]。
称取n-羟乙基-3,3-二甲基-6-硝基吲哚啉螺吡喃0.03g、[(n-c4h9)4n]2[mo6o19]0.1g、eu(no3)3·6h2o0.13g于50ml烧杯中,依次加入3mln,n’-二甲基甲酰胺与9ml异丙醇、搅拌、滴加乙二胺溶液调节反应溶液的ph至8。将烧杯中的溶液转移至容积为25ml聚四氟乙烯内衬的高压不锈钢反应釜中,将反应釜密封后,置于电热恒温鼓风干燥箱中,设定温度为80℃反应3天。待反应结束后,自然冷却至室温,获得橙黄色的长条状晶体即为铕配合物c52h44eu2n16o19。
实施例2
一种具有催化光降解有机染料功能的铕配合物的应用,包括以下步骤:
步骤i、亚甲基蓝溶液的配制:
称取亚甲基蓝(mb)于烧杯中,溶解,然后转移至500ml容量瓶中,定容至刻度线,配制成浓度为10mol/l的亚甲基蓝水溶液,待用;
步骤ii、铕配合物c52h44eu2n16o19的光催化测定:
分别称取2mg、5mg、8mg的铕配合物c52h44eu2n16o19于烧杯中,加入40ml亚甲基蓝水溶液,在黑暗中用磁力搅拌器搅拌30min,直至达到吸附-去吸附过程平衡。在光照下进行光催化反应。每隔30min静置后移取上层清液4ml于试管中,用所得澄清溶液进行紫外光谱检测,紫外检测波长范围是500到800nm。
实施例3
一种具有催化光降解有机染料功能的铕配合物的应用,包括以下步骤:
步骤i、甲基橙溶液的配制:
称取甲基橙(mo)于烧杯中,溶解,然后转移至500ml容量瓶中,定容至刻度线,配制成浓度为10mol/l的甲基橙水溶液,待用;
步骤ii、铕配合物c52h44eu2n16o19的光催化测定:
分别称取2mg、5mg、8mg的铕配合物c52h44eu2n16o19于烧杯中,加入40ml甲基橙水溶液,在黑暗中用磁力搅拌器搅拌30min,直至达到吸附-去吸附过程平衡。在光照下进行光催化反应。每隔30min静置后移取上层清液4ml于试管中,用所得澄清溶液进行紫外光谱检测,紫外检测波长范围是350到650nm。
实施例4
一种具有催化光降解有机染料功能的铕配合物的应用,包括以下步骤:
步骤i、盐酸副玫瑰苯胺溶液的配制:
称取盐酸副玫瑰苯胺(ph)于烧杯中,溶解,转移至500ml容量瓶,定容至刻度线,配制成浓度为10mol/l的盐酸副玫瑰苯胺水溶液,待用;
步骤ii、铕配合物c52h44eu2n16o19的光催化测定:
分别称取2mg、5mg、8mg的铕配合物c52h44eu2n16o19于烧杯中,加入40ml盐酸副玫瑰苯胺水溶液,在黑暗中用磁力搅拌器搅拌30min,直至达到吸附-去吸附过程平衡。在光照下进行光催化反应。每隔30min静置后移取上层清液4ml于试管中,用所得澄清溶液进行紫外光谱检测,紫外检测波长范围是350到650nm。
实施例5
一种具有催化光降解有机染料功能的铕配合物的应用,包括以下步骤:
步骤i、罗丹明b溶液的配制:
称取罗丹明b(rhb)于烧杯中,溶解,然后转移至500ml容量瓶中,定容至刻度线,配制成浓度为10mol/l的甲基橙水溶液,待用;
步骤ii、铕配合物c52h44eu2n16o19的光催化测定:
分别称取2mg、5mg、8mg的铕配合物c52h44eu2n16o19于烧杯中,加入40ml罗丹明b水溶液,在黑暗中用磁力搅拌器搅拌30min,直至达到吸附-去吸附过程平衡。在光照下进行光催化反应。每隔30min静置后移取上层清液4ml于试管中,用所得澄清溶液进行紫外光谱检测,紫外检测波长范围是400到700nm。
为了进一步证明本发明的有益效果以更好地理解本发明,下面通过以下测定试验进一步阐明本发明所述的铕配合物c52h44eu2n16o19具有的性质及应用性能,但不可理解为对本发明的限定,对于本领域的技术人员根据上述发明内容所作的其他测定实验得到的产品性质及根据上述性质进行的应用,也视为落在本发明的保护范围内。
实验例1
目标产物纯度表征:
采用具有d/tex超级衍射仪和以cukα
实验例2
目标产物晶体结构表征:
在室温下,通过显微镜观察选取合适大小的实施例1的目标产物晶体,在室温下进行x-射线衍射实验。晶体的x-射线衍射数据在oxforddiffractiongeminirultra衍射仪上收集,用经石墨单色器单色化的cu-kα射线
表1铕配合物c52h44eu2n16o19的晶体学数据和结构参数
表2铕配合物c52h44eu2n16o19的主要的键长
表3铕配合物c52h44eu2n16o19的主要的键角(o)
实验例3
铕配合物c52h44eu2n16o19的红外光谱分析:
如图2所示,在实施例1的目标产物铕配合物c52h44eu2n16o19中,在3444.72cm-1处为一宽吸收峰,应是水分子中羟基的伸缩振动吸收峰。在3327.08cm-1处为一尖锐吸收峰,应是n—h的伸缩振动吸收峰。在3282.73cm-1处是苯环上不饱和的c—h的伸缩振动吸收峰,2903.09cm-1处是饱和碳上的c—h的伸缩振动吸收峰。在1635.82cm-1、1598.22cm-1、1553.38cm-1处主要是硝基中的n=o的伸缩振动吸收峰和苯环上骨架的振动峰(σc=c)。在1311.36cm-1处为一尖锐吸收峰,应是c—n的伸缩振动吸收峰。在1475-1000cm-1区间中的1238.56cm-1、1099.23cm-1处是c—c单键骨架的振动吸收峰。在1000-650cm-1区间内主要是苯环上的c—h面外弯曲振动区,其中833.10cm-1处是苯环对二取代的c—h面外弯曲振动峰,而760.30cm-1处则是苯环邻二取代的c—h面外弯曲振动峰。通过文献得知,eu—o键、eu—n键的伸缩振动吸收峰分别为474cm-1、317cm-1,故468.39cm-1处则是eu—o键的伸缩振动吸收峰,而图未出现明显的eu—n键的伸缩振动吸收峰,可能是在红外光谱测试时所设定的波数范围不包括eu—n键的伸缩振动吸收峰,故未在图中所体现出eu—n键的伸缩振动吸收峰。
实验例4
铕配合物c52h44eu2n16o19的紫外可见吸收光谱测定
利用tu-1901双光束紫外可见分光光度计对实施例1中的n-羟乙基-3,3-二甲基-6-硝基吲哚啉螺吡喃分子及目标产物铕配合物c52h44eu2n16o19进行紫外可见吸收光谱测定。将仪器预热30min,并设置实验测试仪器的参数与条件选择。在完成实验测试条件的设置后,分别准确称取0.005g螺吡喃分子与晶体溶于装有适量dmf溶剂的10ml样品管中,摇匀使之完全溶解并静置。同时以dmf溶剂为参比,分别对螺吡喃分子和铕配合物进行紫外可见吸收光谱的测定。
螺吡喃分子与铕配合物的紫外可见吸收光谱实验结果如图3所示,在265nm处有一个吸收峰,主要是螺吡喃分子和铕配合物中苯环的π-π*所产生的吸收峰,为芳香族化合物的特征吸收峰。而在351nm和389nm处均存在着吸收峰,应由螺吡喃分子和铕配合物中与苯并吡喃环相连的硝基的n→π*跃迁所产生,同时也表明螺吡喃分子和铕配合物中可能存在着较大的共轭体系。
试验例5
铕配合物c52h44eu2n16o19的荧光性质测试
在稀土eu(ⅲ)化合物中,配体的类型对eu(ⅲ)化合物的发光性能往往具有关键性的影响。稀土eu(ⅲ)化合物的发射特征荧光有3步的过程,即
(1)配体吸收激发光。
(2)所吸收的能量转移给eu3+。
(3)eu3+发射荧光。
根据dexter固体敏化理论,稀土离子发光效率通常是取决于配体三重态能级与稀土离子激发态能级的匹配程度,即有机配体的三重态能级与稀土离子激发态能级间存在最佳匹配。在室温的条件下,若是配体的三重态能级与eu3+的能级(5d0)之差小于4000cm-1,则此时的有机配体敏化程度最大。
在实验时,于室温下设定狭缝宽度为(5nm,10nm),以365nm为激发波长,对化合物进行荧光测试,其结果如图4所示。由图4可知,eu(iii)的特征光谱在578nm,590nm,610nm,649nm和703nm处的发射峰分别对应于5do→7fo,5do→7f1,5do→7f2,5do→7f3和5do→7f4的电子跃迁(如图5所示)。其中,对应于5do→7f0电子跃迁的578nm处的发射峰峰形无分裂现象,表明在化合物中eu3+与配体可能是存在着一种配位模式,即所合成的化合物的纯度较高。在590nm处的发射峰是由5do→7f1磁偶极跃迁产生的,此处的发射强度很少受eu3+配位环境的影响。而在610nm处是eu3+的特征发射峰是归属于5do→7f2电偶极跃迁,此时发射出红色的荧光,半峰宽只有几纳米,具有极高单色性与发光强度,但发光强度则受eu3+配位环境的影响较大。此外,图5中的化合物的5do→7f2发射峰的强度远大于5do→7f1发射峰的强度,表明此化合物的结构对称性并不高。
试验例6
铕配合物c52h44eu2n16o19的光降解测定
利用tu-1901双光束紫外可见分光光度计对实施例2~5光催化降解应用中的铕配合物c52h44eu2n16o19的光降解性能进行表征,图6~21是铕配合物c52h44eu2n16o19对亚甲基蓝溶液(mb)、甲基橙溶液(mo)、盐酸副玫瑰苯胺溶液(ph)和罗丹明b溶液(rhb)的光催化降解紫外吸收光谱图。可以看到,在铕配合物c52h44eu2n16o19加入量为0.05-0.20g·l-1的范围内,随着铕配合物用量的增加,铕配合物对盐酸副玫瑰苯胺(ph)、亚甲基蓝溶液(mb)与甲基橙溶液(mo)的光催化降解率相应增大。这是由于当铕配合物的浓度较低时,对铕配合物的吸附量较少而不能充分进行光降解实验,从而使得有机染料降解率较低。而在图19中,当铕配合物用量为5mg时,铕配合物对罗丹明b溶液(rhb)的光催化降解率最高,可能是因为铕配合物光催化降解罗丹明b溶液时,对铕配合物的吸附量达到相对饱和状态,进而使得罗丹明b溶液降解率达到最大。在有机染料溶液的光催化降解测定实验中,不同的种类有机染料溶液对其光催化降解性能的影响也存在着明显的差异。实验中所使用的有机染料溶液的浓度均为10mol·l-1,其余的实验条件均相同。可以看到,在相同用量的铕配合物的条件下,不同种类有机染料的光催化降解率的比较结果为:亚甲基蓝>盐酸副玫瑰苯胺>罗丹明b>甲基橙。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!