一种非负载型催化剂及三氟氯乙烯或三氟乙烯的制备方法与流程
本发明属于化合物制备领域,涉及三氟氯乙烯和三氟乙烯的制备,具体涉及一种非负载型催化剂及三氟氯乙烯或三氟乙烯的制备方法。
背景技术:
三氟氯乙烯和三氟乙烯都是重要的含氟聚合单体,可制备一系列氟涂料、氟树脂、氟橡胶及氟氯润滑油等。同时,三氟氯乙烯也是一种重要的含氟中间体,可以制备下游产品如三氟乙烯、三氟溴乙烯、六氟丁二烯、三氟苯乙烯、氟溴油等。
传统合成三氟氯乙烯采用1,1,2-三氟-2,2,1-三氯乙烷(cfc-113)锌粉法还原工艺,该工艺为间歇釜式生产,生产设备庞大,效率较低,三氟氯乙烯的生产速率难以控制,并伴有许多副产物的产生(包括二氟乙烯、三氟乙烯、二氟氯乙烯等)。针对金属锌粉还原脱氯工艺所存在的诸多问题,多年来alliedchemistry、uccc、大金、苏威、日本哈龙、大连振邦等众多国内外企业提出了cfc-113催化加氢脱氯制备三氟氯乙烯的工艺新方法。中国专利cn1065261.a和cn1351903公开的加氢脱氯用催化剂都是以贵金属为主催化剂,同时添加其他金属作为助剂,因为贵金属催化剂价格昂贵,因而导致生产成本增加。
美国专利us5089454报道了以活性炭、氧化铝、氧化钛等材料为载体,碱金属和碱土金属盐的一种或多种为助剂,以viii族金属为催化剂活性组分,当反应温度为200~300℃,三氟氯乙烯的转化率在40%左右。在以贵金属为主要活性组分的催化剂中,反应过程中因为催化剂活性高而导致的三氟氯乙烯进一步脱氯产物三氟乙烯选择性不好控制。因此,非常由必要进行能用于cfc-113加氢脱氯的高活性、高选择性催化剂的开发。
目前用于cfc-113催化加氢脱氯制备三氟氯乙烯和三氟乙烯的催化剂价格昂贵或者原料转化率不高等问题。基于此,为应对日益严峻的环保形势和工业应用,迫切需要设计制备低温高活性、环保催化剂用于cfc-113气相选择加氢脱氯制备三氟氯乙烯。
技术实现要素:
针对现有技术存在的不足,本发明的目的在于,提供一种非负载型催化剂及三氟氯乙烯或三氟乙烯的制备方法,解决现有技术中的催化剂难以兼顾原料的转化率、选择性和成本的技术问题。
为了解决上述技术问题,本发明采用如下技术方案予以实现:
一种非负载型催化剂,所述的非负载型催化剂包括含硫体相mop,助剂为氯化铜、氯化钯和氯铂酸中的一种以上组合;
含硫体相mop中,mop为金属钼与非金属磷结合而成具有晶体结构的磷化钼共价化合物,钼与磷的摩尔比为0.3~3;
含硫体相mop中,钼与硫的摩尔比为0.5~2.7;
助剂中金属与钼的摩尔比为0.01~0.7。
本发明还具有如下技术特征:
本发明还保护一种非负载型催化剂的制备方法,所述的非负载型催化剂采用如上所述的非负载型催化剂;
该制备方法具体包括以下步骤:
步骤一,硫化铵与三氯化磷反应制备(nh4)4p2s6;
步骤二,钼酸铵和助剂与氨水反应制备混合溶液;
步骤三,(nh4)4p2s6与步骤二制得得混合溶液反应制备前体盐;
步骤四,氢气还原前体盐制备非负载型催化剂。
步骤一的具体过程为:按照(nh4)2s与三氯化磷摩尔比为1/9~6的计量比称取(nh4)2s,于20℃以下滴加入质量浓度95.5%的三氯化磷水溶液,然后于室温充分反应0.5~2小时制备(nh4)4p2s6。
步骤二的具体过程为:按照mo/p摩尔比为1,助剂中金属/mo摩尔比为0.1的计量比称取钼酸铵和助剂,加入氨水,室温搅拌0.1~1小时得到混合溶液。
步骤三的具体过程为:(nh4)4p2s6与步骤二制得的混合溶液反应制备前体盐:将步骤二中制备的混合溶液加入到步骤一制备的(nh4)4p2s6中,充分混合,于20℃以下搅拌0.5~4小时,然后用乙醇/水溶液洗涤并干燥产物,制备前体盐固体。
步骤四的具体过程为:步骤三制备的前体盐置于管式反应器中,在h2气氛中还原4小时,反应压力0.1~1.2mpa,h2气流量50~120ml/min、反应温度400~600℃,反应时间2~5小时,制得目标催化剂,即非负载型催化剂。
优选的,步骤四中,反应压力0.1mpa,h2气流量100ml/min,温度450℃。
本发明还保护一种三氟氯乙烯或三氟乙烯的制备方法,该方法以1,1,2-三氟-2,2,1-三氯乙烷为原料,该方法采用如上所述的非负载型催化剂作为催化剂,气相选择加氢脱氯制备三氟氯乙烯或三氟乙烯。
反应温度为100~300℃,反应压力为0.1~2.0mpa,接触时间为2~20s,原料氢气与1,1,2-三氟-2,2,1-三氯乙烷的摩尔比为1/1~5/1。
本发明与现有技术相比,具有如下技术效果:
(ⅰ)本发明的催化剂为非负载型催化剂,环境友好、低温活性高。
(ⅱ)本发明的催化剂的原料成本低、绿色环保、高温抗烧结能力好。
(ⅲ)现有技术中,以viii族金属为催化剂活性组分,当反应温度为200~300℃,三氟氯乙烯的转化率在40%左右。本发明提供的催化剂用于cfc-113选择加氢脱氯反应工艺,通过在催化剂中添加不同的金属助剂,可实现在240℃反应时,cfc-113转化率达到95%,同时三氟氯乙烯选择性达到92%;调控催化剂组成,保证原料转化率89%~92%情况下,实现三氟乙烯选择性达到91%~93%。
附图说明
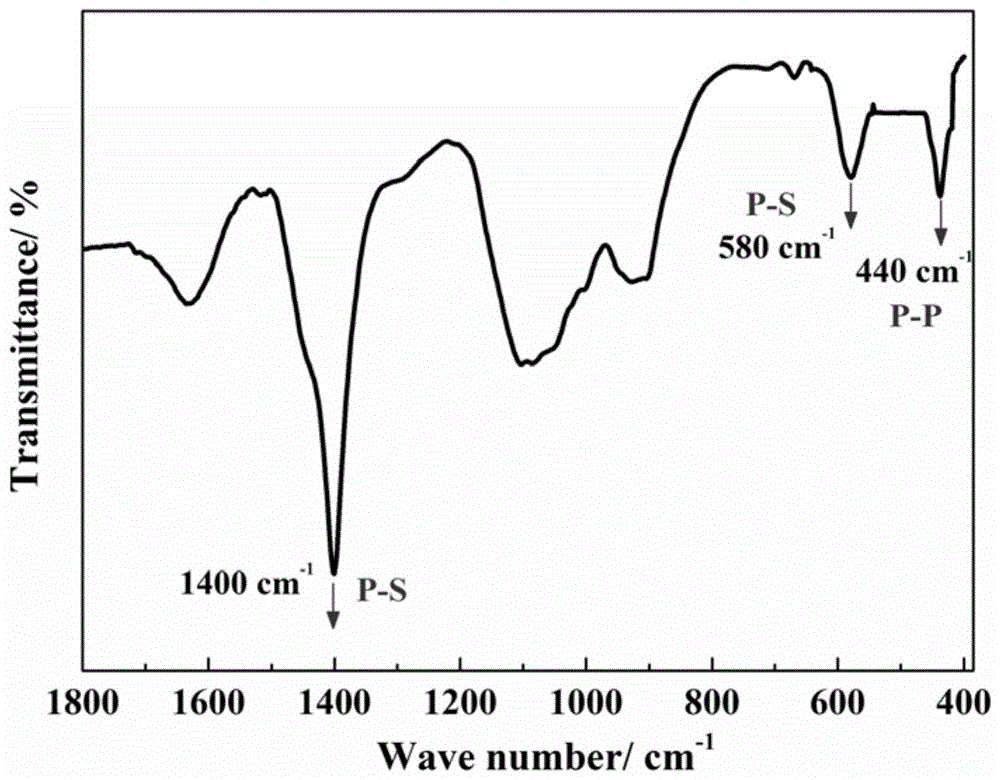
图1是实施例1制得的催化剂a的ir谱图。
图2是实施例1制得的催化剂a的xrd谱图。
图3是实施例1制得的催化剂a的xps谱图。
图4是产物三氟氯乙烯的gc-ms图谱。
图5是实施例2制得的催化剂b的ir谱图。
图6是实施例2制得的催化剂b的xrd谱图。
图7是实施例2制得的催化剂b的xps谱图。
图8是产物三氟乙烯的gc-ms图谱。
图9是实施例3制得的催化剂c的ir谱图。
图10是实施例3制得的催化剂c的xrd谱图。
图11是实施例3制得的催化剂c的xps谱图。
以下结合实施例对本发明的具体内容作进一步详细解释说明。
具体实施方式
本发明的含硫体相mop中,硫主要以两种形式存在:一种是硫化物中的硫,即s2-,另一种是硫醇类化合物中硫物种。
本发明的非负载型催化剂性能测试方法为:量取5ml非负载型催化剂转入固定床管式反应器中,非负载型催化剂床层温度到达240℃后通入cfc-113和氢气,接触时间为8s,h2压力0.2mpa,h2/cfc-113摩尔比为2。运行8h后产物经水、碱洗吸收氟化氢、氯化氢后进气相色谱仪进行分析,采用面积归一化法计算cfc-113的转化率及目标产物选择性。
以下给出本发明的具体实施例,需要说明的是本发明并不局限于以下具体实施例,凡在本申请技术方案基础上做的等同变换均落入本发明的保护范围。
实施例1:
本实施例给出一种的非负载型催化剂,所述的非负载型催化剂包括含硫体相mop,助剂为氯化铜;
含硫体相mop中,mop为金属钼与非金属磷结合而成具有晶体结构的磷化钼共价化合物,钼与磷的摩尔比为1;
含硫体相mop中,钼与硫的摩尔比为0.5;
助剂金属铜与钼的摩尔比为0.7。
本实施例的非负载型催化剂的制备方法,该制备方法具体包括以下步骤:
步骤一,硫化铵与三氯化磷反应制备(nh4)4p2s6;
步骤一的具体过程为:按照(nh4)2s与三氯化磷摩尔比为2的计量比称取(nh4)2s,于20℃以下滴加入质量浓度95.5%的三氯化磷水溶液,然后于室温充分反应0.5~2小时制备(nh4)4p2s6。
步骤二,钼酸铵和助剂金属盐与氨水反应制备混合溶液;
步骤二的具体过程为:按照mo/p摩尔比为1,助剂金属cu/mo摩尔比为0.7的计量比称取钼酸铵和氯化铜,加入氨水,室温搅拌0.1~1小时得到混合溶液。
步骤三,(nh4)4p2s6与步骤二制得得混合溶液反应制备前体盐;
步骤三的具体过程为:(nh4)4p2s6与步骤二制得的混合溶液反应制备前体盐:将步骤二中制备的混合溶液加入到步骤一制备的(nh4)4p2s6中,充分混合,于20℃以下搅拌0.5~4小时,然后用乙醇/水溶液洗涤并干燥产物,制备前体盐固体。
步骤四,氢气还原前体盐制备非负载型催化剂。
步骤四的具体过程为:步骤三制备的前体盐置于管式反应器中,在h2气氛中还原4小时,反应压力0.1~1.2mpa,h2气流量50~120ml/min、反应温度400~600℃,反应时间2~5小时,制得目标催化剂,即非负载型催化剂。
本实施例制得的非负载型催化剂的样品命名为a。
产品表征:
本实施例制得的催化剂a的ir谱图如图1所示,本实施例制得的催化剂a的xrd谱图如图2所示,本实施例制得的催化剂a的xps谱图如图3所示。
从图1中可知,位于580和1400cm-1的两红外吸收峰,对应于p-s键伸缩振动,而位于440cm-1的红外吸收峰,对应于p-p键伸缩振动。
从图2中可知,样品主要晶相为mop(pdf24-0771),2θ在28°、32°、43°以及57°左右处的衍射峰,分别对应mop的(001)、(100)、(101)和(110)晶面。其中还含有cu3p(pdf71-2261)的晶相,2θ在36°、39°、41.6°、45.1°以及46.1°左右处的衍射峰,分别对应cu3p的(112)、(202)、(211)、(300)和(113)晶面。
从图3中可知,催化剂表面含有硫,根据特征峰位置,主要的硫物种为硫化物(s2-)和硫醇类化合物中硫。
因此,本实施例制得的催化剂a即为本申请中所要制得的目标非负载型催化剂。
性能测试:
催化剂a用于cfc-113加氢脱氯制备三氟氯乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:240℃下原料转化率为95%,三氟氯乙烯选择性为92%。图4是产物三氟氯乙烯的gc-ms图谱,质谱结果与标准谱图匹配度高,图4质谱结果及其峰值归属如下:存在m/z=116为分子离子峰,m/z=85为cf2=cfcl脱cf后离子峰,m/z=69为cf2=cfcl脱ccl后离子峰,m/z=47为碎片ccl的离子峰,m/z=31为碎片cf的离子峰。从图4中可以看出,该化合物为三氟氯乙烯。
对比例1:
将催化剂a中的助剂去掉,只保留含硫体相mop,然后将该催化剂用于cfc-113加氢脱氯制备三氟氯乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:在不添加任何其它试剂情况下,cfc-113转化率为69%,三氟氯乙烯的选择性分别为75%,催化剂对反应原料转化率不高,同时单一产物的选择性低,不利于进一步的分离提纯。
实施例2:
本实施例给出一种的非负载型催化剂,所述的非负载型催化剂包括含硫体相mop,助剂为氯化钯;
含硫体相mop中,mop为金属钼与非金属磷结合而成具有晶体结构的磷化钼共价化合物,钼与磷的摩尔比为0.3;
含硫体相mop中,钼与硫的摩尔比为1;
助剂金属钯与钼的摩尔比为0.01。
本实施例的非负载型催化剂的制备方法,该制备方法具体包括以下步骤:
步骤一,硫化铵与三氯化磷反应制备(nh4)4p2s6;
步骤一的具体过程为:按照(nh4)2s与三氯化磷摩尔比为0.3的计量比称取(nh4)2s,于20℃以下滴加入质量浓度95.5%的三氯化磷水溶液,然后于室温充分反应0.5~2小时制备(nh4)4p2s6。
步骤二,钼酸铵和助剂金属盐与氨水反应制备混合溶液;
步骤二的具体过程为:按照mo/p摩尔比为0.3,助剂金属pd/mo摩尔比为0.01的计量比称取钼酸铵和氯化钯,加入氨水,室温搅拌0.1~1小时得到混合溶液。
步骤三,(nh4)4p2s6与步骤二制得得混合溶液反应制备前体盐;
步骤三的具体过程为:(nh4)4p2s6与步骤二制得的混合溶液反应制备前体盐:将步骤二中制备的混合溶液加入到步骤一制备的(nh4)4p2s6中,充分混合,于20℃以下搅拌0.5~4小时,然后用乙醇/水溶液洗涤并干燥产物,制备前体盐固体。
步骤四,氢气还原前体盐制备非负载型催化剂。
步骤四的具体过程为:步骤三制备的前体盐置于管式反应器中,在h2气氛中还原4小时,反应压力0.1~1.2mpa,h2气流量50~120ml/min、反应温度400~600℃,反应时间2~5小时,制得目标催化剂,即非负载型催化剂。
本实施例制得的非负载型催化剂的样品命名为b。
产品表征:
本实施例制得的催化剂b的ir谱图如图5所示,本实施例制得的催化剂b的xrd谱图如图6所示,本实施例制得的催化剂b的xps谱图如图7所示。
从图5中可知,位于580和1400cm-1的两红外吸收峰,对应于p-s键伸缩振动,而位于440cm-1的红外吸收峰,对应于p-p键伸缩振动。
从图6中可知,样品晶相为mop(pdf24-0771),2θ在28°、32°、43°以及57°左右处的衍射峰,分别对应mop的(001)、(100)、(101)和(110)晶面。
从图7中可知,催化剂表面含有硫,根据特征峰位置,主要的硫物种为硫化物(s2-)和硫醇类化合物中硫。
因此,本实施例制得的催化剂b即为本申请中所要制得的目标非负载型催化剂。
性能测试:
催化剂b用于cfc-113加氢脱氯制备三氟乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:240℃下原料转化率为92%,三氟乙烯选择性为93%。图8是产物三氟乙烯的gc-ms图谱,质谱结果与标准谱图匹配度高,图8质谱结果及其峰值归属如下:存在m/z=82为分子离子峰,m/z=63为cf2=chf脱f后离子峰,m/z=51为cf2=chf脱cf后离子峰,m/z=31为碎片cf的离子峰。从图8中可以看出,该化合物为三氟乙烯。
对比例2:
将催化剂b中的助剂去掉,只保留含硫体相mop,然后将该催化剂用于cfc-113加氢脱氯制备三氟氯乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:在不添加任何其它试剂情况下,cfc-113转化率为69%,三氟乙烯的选择性分别为25%,催化剂对反应原料转化率不高,同时单一产物的选择性低,不利于进一步的分离提纯。
实施例3:
本实施例给出一种的非负载型催化剂,所述的非负载型催化剂包括含硫体相mop,助剂为氯铂酸;
含硫体相mop中,mop为金属钼与非金属磷结合而成具有晶体结构的磷化钼共价化合物,钼与磷的摩尔比为3;
含硫体相mop中,钼与硫的摩尔比为2.7;
助剂金属铂与钼的摩尔比为0.3。
本实施例的非负载型催化剂的制备方法,该制备方法具体包括以下步骤:
步骤一,硫化铵与三氯化磷反应制备(nh4)4p2s6;
步骤一的具体过程为:按照(nh4)2s与三氯化磷摩尔比为10/9的计量比称取(nh4)2s,于20℃以下滴加入质量浓度95.5%的三氯化磷水溶液,然后于室温充分反应0.5~2小时制备(nh4)4p2s6。
步骤二,钼酸铵和助剂金属盐与氨水反应制备混合溶液;
步骤二的具体过程为:按照mo/p摩尔比为3,助剂金属pt/mo摩尔比为0.3的计量比称取钼酸铵和氯铂酸,加入氨水,室温搅拌0.1~1小时得到混合溶液。
步骤三,(nh4)4p2s6与步骤二制得得混合溶液反应制备前体盐;
步骤三的具体过程为:(nh4)4p2s6与步骤二制得的混合溶液反应制备前体盐:将步骤二中制备的混合溶液加入到步骤一制备的(nh4)4p2s6中,充分混合,于20℃以下搅拌0.5~4小时,然后用乙醇/水溶液洗涤并干燥产物,制备前体盐固体。
步骤四,氢气还原前体盐制备非负载型催化剂。
步骤四的具体过程为:步骤三制备的前体盐置于管式反应器中,在h2气氛中还原4小时,反应压力0.1~1.2mpa,h2气流量50~120ml/min、反应温度400~600℃,反应时间2~5小时,制得目标催化剂,即非负载型催化剂。
本实施例制得的非负载型催化剂的样品命名为c。
产品表征:
本实施例制得的催化剂c的ir谱图如图9所示,本实施例制得的催化剂c的xrd谱图如图10所示,本实施例制得的催化剂11的xps谱图如图9所示。
从图9中可知,位于580和1400cm-1的两红外吸收峰,对应于p-s键伸缩振动,而位于440cm-1的红外吸收峰,对应于p-p键伸缩振动。
从图10中可知,样品晶相为mop(pdf24-0771),2θ在28°、32°、43°以及57°左右处的衍射峰,分别对应mop的(001)、(100)、(101)和(110)晶面。
从图11中可知,催化剂表面含有硫,根据特征峰位置,主要的硫物种为硫化物(s2-)和硫醇类化合物中硫。
因此,本实施例制得的催化剂c即为本申请中所要制得的目标非负载型催化剂。
性能测试:催化剂c用于cfc-113加氢脱氯制备三氟乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:240℃下原料转化率为89%,三氟乙烯选择性为91%。本实施例制得的产物三氟乙烯的gc-ms图谱与图8基本相同。
对比例3:
将催化剂c中的助剂去掉,只保留含硫体相mop,然后将该催化剂用于cfc-113加氢脱氯制备三氟乙烯反应中,采用本发明的非负载型催化剂性能测试方法可得:在不添加任何其它试剂情况下,cfc-113转化率为69%,三氟乙烯的选择性分别为25%,催化剂对反应原料转化率不高,同时单一产物的选择性低,不利于进一步的分离提纯。
实施例4至6:
本实施例给出一种制备三氟氯乙烯或三氟乙烯的制备方法,该方法以1,1,2-三氟-2,2,1-三氯乙烷为原料,该方法采用实施例1、实施例2或实施例3所述的的非负载型催化剂作为催化剂,气相选择加氢脱氯制备三氟氯乙烯或三氟乙烯;
具体的反应过程为:量取5ml非负载型催化剂转入固定床管式反应器中,非负载型催化剂床层温度到达240℃后通入cfc-113和氢气,接触时间为8s,h2压力0.2mpa,h2/cfc-113摩尔比为2。运行8h后产物经水、碱洗吸收氟化氢、氯化氢后制得产物。
具体的原料转化率,产物选择性以及产物的gc-ms图谱同于实施例1、实施例2或实施例3。
实施例7:
本实施例给出一种制备三氟氯乙烯或三氟乙烯的制备方法,该方法以1,1,2-三氟-2,2,1-三氯乙烷为原料,该方法采用实施例1、实施例2或实施例3所述的的非负载型催化剂作为催化剂,气相选择加氢脱氯制备三氟氯乙烯或三氟乙烯;
反应温度为100~300℃,反应压力为0.1~2.0mpa,接触时间为2~20s,原料氢气与1,1,2-三氟-2,2,1-三氯乙烷的摩尔比为1/1~5/1。
- 还没有人留言评论。精彩留言会获得点赞!