一种铬铁酸锌(ZnCr的制作方法
一种铬铁酸锌(zncr
2-x
fe
x
o4)尖晶石电催化剂及其制备方法和应用
技术领域
1.本发明涉及一种铬铁酸锌(zncr
2-x
fe
x
o4)尖晶石电催化剂制备方法及在电化学合成氨中的应用。
背景技术:
2.氨是世界上大量生产的化学物质之一,年产量超过1.82亿吨,主要用作合成肥料。氨还具有含氢量高(质量比达17.6%)、能量密度高、易液化等优点,有望成为清洁储氢载体。目前,氨是通过催化haber-bosch过程于高温高压(300~500℃、200~300atm)下由氮气和氢气合成生产,需要大型工厂和高投资,能源消耗约占全球总能耗的1~2%,同时,每年排放量全球1.5%的二氧化碳。因此,实现在常温常压下高效合成氨,是人们梦寐以求的。电化学合成氨以水为氢源,可在常温常压下进行,且可通过太阳能或风能等绿色能源转化的电能驱动,将能够彻底克服haber-bosch法合成氨所面临的涉及能耗、污染以及安全性等方面的问题,被认为是一种潜在的合成氨替代技术。然而,迄今为止,处于核心地位的电催化氮气还原合成氨催化剂受到吸附能线性关系的严重限制,催化活性和选择性仍处于较低水平。对n2吸附强的前过渡金属元素(元素周期表靠左的元素)例如铬(cr)、钒(v)和锰(mn),在合成氨反应气氛中易形成稳定的氮化物相,阻碍了后续加氢步骤,合成氨活性较差;而对n2吸附相对弱的后过渡金属(元素周期表靠右的元素)例如钌(ru)和铁(fe)因具有较为适中的n2吸附能,表现出优异的电催化合成氨活性,但是对氢气吸附较强,无法有效抑制析氢反应(her),选择性不理想,如何突破这种制约关系成为目前电化学合成氨发展的动力学瓶颈。因此,开发出能够打破线性关系限制的催化剂材料,是横亘在科学家面前的一大难题。
3.尖晶石(结构通式为ab2o4)具有立方晶体结构,氧离子呈立方紧密堆积,二价阳离子a占据四次配位的四面体空隙,三价阳离子b占据六次配位的八面体空隙,尖晶石材料a位和b位阳离子种类均可调控,阳离子的分布对尖晶石型材料的性能有重大影响,已被广泛用于磁性材料、微波电介质材料、湿敏和气敏材料和化学催化等领域。文献(河北大学学报(自然科学版),2012,12(6):619-622)采用固相法以zno、fe2o3、cr2o3为原料,经研磨、高温焙烧制备zncr
2-x
fe
x
o4(x=0,0.3,0.5,0.8,1.0)系列尖晶石并用作磁性材料,发现需要1100℃以上的高温才能成功在b位掺入fe
3+
,而且制备的材料纯度不高,含有cr2o3、zno等杂相,影响其进一步的应用。文献(results in physics,2021,28:104622)以硝酸锌、硝酸镍、硝酸铬和硝酸铁粉末为原料,以为甘氨酸燃料采用微波燃烧法制备a位的zn被ni、cr部分取代的ni
0.4
zn
0.6-x
cr
x
fe2o4(0.0≤x≤0.6)系列尖晶石,并用作磁性材料。寻求一种简单可控的b位掺入fe
3+
的纯净zncr
2-x
fe
x
o4合成方法具有重要意义。
技术实现要素:
4.为了解决背景技术中所提到的技术问题,本发明提供一种铬铁酸锌尖晶石电催化剂制备方法及应用,将前过渡金属cr和后过渡金属fe引入到锌基尖晶石八面体空隙,构成
双活性位点体系,制备出一种铬铁酸锌zncr
2-x
fe
x
o4(x=0,0.4,0.8,1.2,1.6,2)电催化剂,通过cr和fe活性位点的协同作用,在一定程度上规避了单一过渡金属上的线性关系限制,实现了氨的高效电催化合成。
5.本发明的技术方案是:一种铬铁酸锌尖晶石电催化剂制备方法,具体步骤为:
6.第一步,按一定比例称取锌酸盐、铬酸盐和铁酸盐溶于溶剂中,然后加入碱溶液调节ph=10~12,然后水热反应,后处理后得到干燥的催化剂前体。
7.锌酸盐、铬酸盐和铁酸盐摩尔比为1:(2-x):x,x大于0小于2;
8.水热合成温度范围在150℃~200℃;
9.第二步,将第一步烘干后获得的催化剂前体在马弗炉中焙烧,自然冷却至室温后研磨获得所需电催化剂;
10.进一步地,在上述技术方案中,第一步中,所述溶剂为去离子水;所述碱溶液为3.0-6.0mol/l氢氧化钠或氢氧化钾水溶液。
11.进一步地,在上述技术方案中,第一步中,水热反应及后处理为将金属前驱体转入带有聚四氟乙烯衬里的水热反应釜中在一定温度下水热反应一定时间,反应后,离心分离去除上清液后获得催化剂前体,用去离子水洗涤3次,真空干燥。
12.进一步地,在上述技术方案中,第一步中,水热合成时间范围在4小时~12小时之间,真空干燥温度范围在60℃~80℃之间。
13.进一步地,在上述技术方案中,第一步中,所述锌酸盐包括氯化锌、硫酸锌、硝酸锌和醋酸锌;铬酸盐包括氯化铬、硫酸铬和硝酸铬;铁酸盐包括氯化铁、硫酸铁和硝酸铁;锌酸盐的浓度为0.05~0.2mol/l。
14.进一步地,在上述技术方案中,第二步中,焙烧温度范围在300℃~500℃之间,焙烧时间范围在0.5小时~3小时之间。
15.进一步地,在上述技术方案中,第一步中,所述锌酸盐、铬酸盐和铁酸盐分别为醋酸锌、氯化铬和氯化铁,三者的摩尔比为1:1.2:0.8,醋酸锌浓度为0.1mol/l,水热合成温度为180℃,水热合成时间为8小时;第二步中,所述焙烧温度为400℃,焙烧时间为1小时。
16.本发明提供上述方法得到的纯净铬铁酸锌尖晶石电催化剂,由通式zncr
2-x
fe
x
o4表示,2-x、x分别表示各元素的原子比,x大于0小于2。
17.进一步地,在上述技术方案中,x选自0.4,0.8,1.2,1.6中的任一个数。
18.本发明提供上述的方法制得的电催化剂在电化学合成氨中应用,具体过程为:将所制备的电催化剂负载到碳毡上作为阴极,以未负载催化剂的碳毡为阳极,以koh水溶液为电解质,以一定流量(200ml/min)向阴极吹高纯氮气,然后接通直流电源在室温常压下恒电压(1.5~2.5v)电解,具有优异的电化学还原氮气产氨性能。
19.本发明具有如下有益效果:(1)催化剂制备所需原料易得,成本低,操作简单,可控性好,易于量产。(2)本发明制备的电化学合成氨的催化剂为纳米铬铁酸锌尖晶石。(3)采用制备的电催化剂,以0.1mol/lkoh水溶液为电解质,以氮气为原料气,电化学合成氨的产氨速率和法拉第效率最高分别可达29.26μg h-1
cm-2
和18.41%。术语“法拉第效率”指实际生成氨与电化学理论生成氨的百分比。
附图说明
20.图1是实施例1制备得到的催化剂xrd图;
21.图2是实施例1制备得到的zncr2o4、zncr
1.2
fe
0.8
o4和znfe2o4催化剂的xps表征结果;
22.图3是实施例1制备得到的zncr
1.2
fe
0.8
o4催化剂tem和hrtem表征结果;
23.图4是实施例1制备得到的zncr2o4、zncr
1.2
fe
0.8
o4和znfe2o4催化剂氮气程序升温脱附结果;
24.图5实施例1制备的催化剂在2.1v下电化学合成氨性能;
25.图6实施例1制备得到的zncr
1.2
fe
0.8
o4催化剂在不同电解电压下合成氨的电流密度;
26.图7实施例1制备得到的zncr
1.2
fe
0.8
o4催化剂在不同电解电压下合成氨的产氨速率和法拉第效率;
27.图8对比例1制备得到的样品xrd图;
28.图9对比例2制备得到的样品xrd图。
具体实施方式
29.下面通过实施例对本发明的上述内容做进一步详细说明。
30.实施例1
31.(1)按化学式zncr
2-x
fe
x
o4(x=0,0.4,0.8,1.2,1.6,2)给出的比例1:(2-x):x称取5mmol的醋酸锌和相应量的氯化铬和氯化铁溶于去离子水中得到6种不同配比的溶液,采用3.0mol/l氢氧化钠水溶液调节溶液的ph=10~12,再加水使溶液总体体积为50ml,然后分别转入带有聚四氟乙烯衬里的6个水热反应釜中在180℃下水热反应8小时,反应后,离心分离去除上清液后获得6种催化剂前体,用去离子水洗涤3次,真空干燥后备用。
32.(2)将烘干后获得的催化剂前体分别在马弗炉中于400℃下焙烧1小时,自然冷却至室温后研磨获得所需电催化剂。
33.对实施例1得到的催化剂进行x-射线衍射(xrd)分析,由图1可以看出,所有样品具有相似的xrd谱图,在2θ=18.4
°
,30.3
°
,35.7
°
,43.4
°
,53.9
°
,57.4
°
和63.1
°
附近具有明显的特征衍射峰,归属于zncr2o4尖晶石标准卡片pdf no.22
–
1107和znfe2o4尖晶石标准卡片pdf no.22
–
1012的(111)、(220)、(311)、(400)、(422)、(511)和(440)晶面,同时没有其他杂峰出现,表明所制备的催化剂具有尖晶石结构,且无杂相。随着fe含量(x值)的增加,所有特征衍射峰均向2θ小角度方向移动。对xrd谱线出现的(311)晶面对应的衍射峰,依据晶面间距公式1/d
2hkl
=(h2+k2+l2)/a2和bragg方程nλ=2d
hkl
sinθ(式中a为晶格常数,h,k和l为晶面miller指数,d
hkl
为(h,k,l)晶面的晶面间距,n为衍射级数,通常n=1,λ为x-射线波长,cu kα1(λ=0.15406nm),θ为衍射角)计算不同组成催化剂的(311)晶面间距,计算表明,当x值从0提高到2时,晶格常数a从逐渐增加至说明fe含量的增加导致晶格膨胀,这主要是由于半径较大的fe
3+
离子取代了尖晶石八面体中心的半径相对小些的所致,也表明八面体中心的cr
3+
部分地被fe
3+
取代,共同形成均匀的、单一相的尖晶石晶体。
34.对实施例1得到的zncr2o4,zncr
1.2
fe
0.8
o4和znfe2o4催化剂的进行x-射线光电子能
谱(xps)表征,图2a中zn 2p谱图在结合能1021.4ev和1044.3ev出现2个峰,同时zn lmm俄歇谱图(图2b)约在~989ev俄歇电子动能处出现异于金属zn的(992ev)谱峰,表明zn呈zn
2+
价态。比较zncr2o4和zncr
1.2
fe
0.8
o4的cr 2p谱图(图2c)可以看出,二者没有明显差异,均在578.8和576.5ev结合能处出现cr 2p
3/2
峰,在588.3和586.2ev结合能处出现cr 2p
1/2
峰,说明cr的价态为cr
3+
。zncr
1.2
fe
0.8
o4与znfe2o4的fe2p谱图类似(图2d),在711.0和713.5ev结合能处出现fe2p
3/2
峰,同时在fe2p
3/2
峰高结合能一侧出现典型的fe
3+
卫星峰,表明fe的价态为fe
3+
。
35.对实施例1得到的zncr
1.2
fe
0.8
o4催化剂进行透射电子显微镜(tem)表征(图3),可以看出,催化剂粒径分布在较窄的范围内,平均粒径约为6.6nm,选区电子衍射(saed)谱图出现明显的(311)、(400)、(422)和(440)晶面的衍射环,说明催化剂为多晶结构;由高分辨率透射电镜(hrtem)表征发现晶粒具有排列规律的间距为0.252nm和0.209nm的晶格条纹,分别归属于(311)和(400)晶面,这与xrd结果相一致。
36.实施例2
37.对实施例1得到的zncr2o4、zncr
1.2
fe
0.8
o4和znfe2o4催化剂的进行氮气程序升温脱附(n
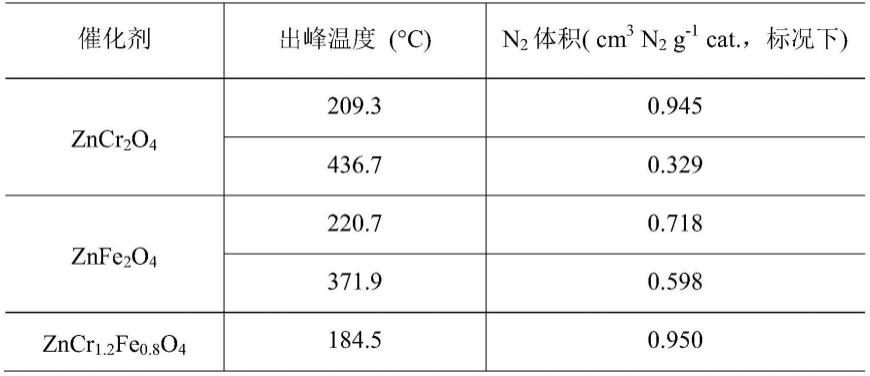
2-tpd)测试,以便确定制备的催化剂对氮气的化学吸附性能,结果见图4和表1。化学吸附有利于拉长氮气n≡n键而活化,但是对于氮气还原产氨来说,要求氮气吸附强度适中,不能太强也不能太弱。由图4可以看出,未引入fe之前,zncr2o4在209.3和436.7℃处出现氮气脱附峰;类似地,znfe2o4在220.7℃和371.9℃处出现明显的脱附峰,表明二者对氮气的化学吸附较强,而引入fe之后的zncr
1.2
fe
0.8
o4的氮气脱附峰温度降低出现在184.5℃,说明引入fe降低了对氮气的化学吸附强度。值得注意的是由表1可以看出zncr
1.2
fe
0.8
o4对氮气的吸附量并没有明显降低,仍为0.950cm3n2/g cat,与zncr2o4在209.3℃处的吸附量(0.945cm3n2/gcat.)几乎相等,表明引入fe改进了催化剂对氮气的吸附性能,解决了纯cr基尖晶石催化剂对氮气吸附过强问题。
38.表1 n
2-tpd峰出峰温度和脱附n2体积
[0039][0040]
实施例3
[0041]
对实施例1制备的zncr
2-x
fe
x
o4(x=0,0.4,0.8,1.2,1.6,2)进行电化学合成氨性能进行测试。将所制备的电催化剂负载到碳毡上作为阴极,以0.1mol/l的koh水溶液为电解质,以碳毡为阳极,以200ml/min的流量向阴极吹氮气,然后接通电源,在2.1v电压下恒电压电解,分析测定电解产生的氨气,结果见图5,可以看出,zncr
1.2
fe
0.8
o4(x=0.8)的产氨速率
和法拉第效率高达29.26μg h-1
cm-2
和8.16%,比zncr2o4(x=0)明显提高,主要是引入fe改进了催化剂对氮气的化学吸附和活化性能,同时znfe2o4(x=2.0)的产氨速率和法拉第效率最低,表明该催化剂作用下可能有更多的her副反应发生。
[0042]
实施例4
[0043]
与实施例1的不同在于电压不同,本实施例考察不同电解电压下电化学合成氨性能。参见图6,图6为zncr
1.2
fe
0.8
o4在不同电解电压下的电流密度曲线,由图可以看出,随着电解的进行,电解电流密度在电解时间内保持稳定,表明在催化剂上有平稳的电化学反应发生。图7为zncr
1.2
fe
0.8
o4电催化产氨性能结果,可以看出,当电压为1.5v时,得到10.08μg h-1
cm-2
的产氨速率和高达18.41%的法拉第效率,随着电压的提高,电解电流几乎线性增加,产氨速率也同步提高,2.1v时获得最大产氨速率为29.26μg h-1
cm-2
,此时法拉第效率为8.16%。
[0044]
实施例5
[0045]
本实施例与实施例1相仿,其不同点在于:步骤(1)仅按比例1:1.2:0.8称取相应量的醋酸锌、氯化铬和氯化铁,水热温度为150℃,其余步骤与实施例1相同,制备zncr
1.2
fe
0.8
o4。2.1v恒电压电解时,产氨速率为28.43μg h-1
cm-2
,法拉第效率为8.09%。
[0046]
对比例1
[0047]
为了与实施例1制备的催化剂(zncr
1.6
fe
0.4
o4)相对比,用co替换cr,按化学式znco
1.6
fe
0.4
o4给出的比例1:1.6:0.4称取5mmol的醋酸锌和相应量的氯化钴和氯化铁溶于去离子水中,其余步骤同实施例1。采用3.0mol/l氢氧化钠水溶液调节溶液的ph=10~12,再加水使溶液总体体积为50ml,然后转入带有聚四氟乙烯衬里水热反应釜中在180℃下水热反应8小时,反应后,离心分离去除上清液后获得催化剂前体,用去离子水洗涤3次,真空干燥。在马弗炉中于400℃下焙烧1小时,自然冷却至室温后研磨获得所需电催化剂。
[0048]
对对比例1得到的样品进行xrd分析,由图8可以看出,制备获得的样品xrd谱图与实施例1中的zncr
1.6
fe
0.4
o4的xrd谱图(见图1中x=0.4)明显不同,在2θ=31.8
°
,34.4
°
,36.3
°
,47.6
°
,56.6
°
,62.9
°
和67.9
°
处出现明显衍射峰,归属于zno(标准卡片pdf no.36
–
1451)。在2θ=18.9
°
,31.2
°
,36.8
°
,44.7
°
,59.3
°
,65.1
°
和63.1
°
处出现明显的特征衍射峰,归属于znco2o4尖晶石(标准卡片pdf no.23
–
1390),这表明对比例1制备的样品中含有zno和znco2o4相。需要注意的是,虽然按照化学式znco
1.6
fe
0.4
o4给出的计量比1:1.6:0.4投加各种原料盐类,但是图8所示xrd谱图除了与zno和znco2o4相吻合的衍射峰外,没有其他杂峰出现,尤其没有出现与znfe2o4尖晶石(pdf no.22
–
1012)相符合的系列特征衍射峰,说明对比例1制备的样品中不含有znfe2o4尖晶石相,原料中加入的氯化铁合成反应后并没有生成相关的任何含铁晶体物种(没有多余的杂峰),或者仅生成了无定形的非晶态fe基物质。因此,对比例1制备的样品晶体结构不是纯净的znco
1.6
fe
0.4
o4尖晶石,而是zno和znco2o4尖晶石的混合物。
[0049]
对比例1得到的样品在2.1v下恒电压电解时,产氨速率为6.38μg h-1
cm-2
,远低于实施例1制备的zncr
1.6
fe
0.4
o4催化剂(20.83μg h-1
cm-2
);法拉第效率为4.91%,也低于实施例1制备的zncr
1.6
fe
0.4
o4催化剂(5.07%)。
[0050]
对比例2
[0051]
为了与实施例1合成的催化剂(zncr
1.6
fe
0.4
o4)相对比,用co替换fe,按化学式
zncr
1.6
co
0.4
o4给出的比例1:1.6:0.4称取5mmol的醋酸锌和相应量的氯化铬和氯化钴溶于去离子水中,其余步骤同实施例1。
[0052]
图9为对比例2制备样品的xrd谱图,由图9可以看出,制备获得的样品xrd谱图与实施例1中的zncr
1.6
fe
0.4
o4的xrd谱图(见图1中x=0.4)类似,在2θ=18.4
°
,30.3
°
,35.7
°
,43.4
°
,57.4
°
和63.1
°
附近出现明显的特征衍射峰,归属于zncr2o4尖晶石(标准卡片pdf no.22
–
1107)和znco2o4尖晶石(标准卡片pdf no.23
–
1390),并且没有其他杂峰出现,说明对比例2制备的样品具有zncr
1.6
co
0.4
o4尖晶石结构,且无杂相。2.1v恒电压电解时,产氨速率为14.34μg h-1
cm-2
,法拉第效率为4.26%,均低于实施例1制备的zncr
1.6
fe
0.4
o4催化剂。
[0053]
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1