一种青霉素皮试冻干粉剂的质量检测方法与流程
本发明涉及药学和药物分析
技术领域:
,具体涉及一种青霉素皮试冻干粉剂的质量检测方法。
背景技术:
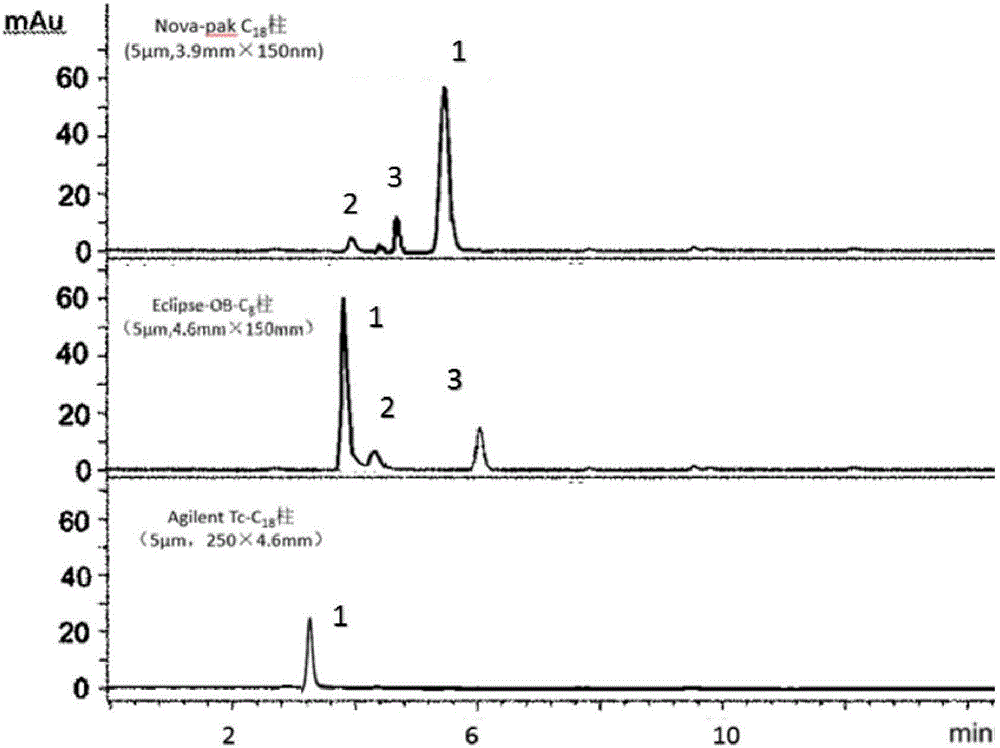
:青霉素类(Penicilins)药物是一类β-内酞胺类抗生素的总称,因其高效、低毒是目前医学临床上应用最受青睐的药物之一,虽然青霉素类药物本身对机体没有很强毒性,但容易引起药物过敏反应,是各类过敏药物之最。青霉素过敏反应的原因主要是青霉素的降解产物青霉烯酸、青霉噻唑酸以及其聚合物,这些物质作为半抗原进入人体后与蛋白质或多肽分子结合成全抗原,其中最主要的是青霉噻唑蛋白,它是引起大多数人过敏反应的主要原因。因此,对青霉素类药物在临床应用时必须先进行皮肤过敏试验,历年来都是我国基础护理常规操作之一,中国药典2010版中也明确规定,注射或口服青霉素类药物之前,均需进行皮试,皮试药液的浓度为500单位/ml,阴性反应者方能使用该类药物,且用药过程中药物的批号有更换时,也必须重做皮内试验。资料表明,青霉素皮试剂,从第一代的青霉素G(PG),到第二代的青霉噻唑酰-多赖氨酸(PLL)、青霉噻唑羧酸盐(BPNCO),一定程度上解决了过敏反应,但仍存在“假阳性反应”,特别是婴幼儿,其假阳性率高达10.9%(李亚玲.《青霉素皮试假阳性30例分析》.中国医药导报.2008.5(17):137),给分析、判断皮试结果带来了很大的干扰,究其原因在于:①青霉素类药物的分子结构决定了青霉素类水剂不稳定,易降解、失效,一般新配置的皮试液要冷藏保存,使用期一般为一周以内,放置时间过长易分解为青霉噻唑和青霉烯酸等过敏物质;且由于放置时间过长,多次抽取,空气气泡来回倒入也会造成污染,尤其是在夏季皮试后,易引起假阳性反应;②受剂型的限制,需要通过多次抽取、混合逐步配制成浓度为500单位/ml的皮试液(药典规定)方可使用,该配制方法会造成青霉素溶解不充分,影响皮试结果的判定,对婴幼儿来说会明显增加皮试结果的假阳性率,延误治疗。再者,目前国内市场上流通着不同品种、不同厂家、不同工艺流程、不同批号的青霉素类药物,导致所含的致敏物的种类和含量不同。为此,需要开发出一种更合适的青霉素皮试剂型(传统的青霉素皮试剂,多为注射剂或混悬针剂),以解决现有皮试剂的制剂稳定性差、易出现假阳性反应、临床用药存在安全隐患等缺点。公开号为CN103861125A的中国发明专利,提供了新型青霉素皮试冻干粉剂及制备工艺,其皮试冻干粉剂每瓶或每支内主要含有青霉素钠盐1000-10000单位,其制备工艺是通过将原料药青霉素钠盐用注射用水稀释后,再加入特定比例的赋形剂,通过优化的工艺制成冻干粉剂。临床使用时,只需注入一定量的生理盐水,混匀,即可配制成浓度为500单位/ml的皮试液。该专利提供的皮试冻干粉剂,相比于传统的医用皮试剂,具有制剂稳定性好、降低假阳性率的优点,是一种用青霉素类皮试冻干粉剂来替代传统皮试注射剂的药物开发思路,但其不足之处在于:专利中并未涉及皮试冻干粉剂的有关物质测定、含量测定的方法,而在制剂生产中,如何快速、准确的检测有关物质的含量,将其控制在药品标准范围内,对青霉素皮试冻干粉剂的产业化与临床用药安全有着非常重要的作用。令人遗憾的是,在《中国药典》(2015版)中尚未收录青霉素类皮试冻干粉剂的质量检测方法,药典只提供了青霉素钠与注射用青霉素钠的检测方法,其色谱条件为:色谱柱:C18柱;流动相A:磷酸二氢钾-甲醇(72:14),流动相B:乙腈,流动相A:流动相B=86.5:13.5;流速:1mL/min;检测波长:225nm,等度洗脱。发明人将药典方法应用于青霉素钠皮试冻干粉剂的质量检测时,发现存在如下技术缺陷:①采用药典方法,其色谱体系的主峰保留时间较短,辅料峰与杂质峰有重叠、分离度差,积分有干扰;②药典方法的检测体系稳定性不佳,检测结果重现性差,不具有稳定性指示作用。发明人经检索,发现涉及青霉素皮试剂质量检测的,相关文献如下:文献《旋光法测定青霉素皮试注射液的含量》(倪红辉.中国药业.2008.17(23):33)中,公开了一种青霉素皮试注射液的含量测定方法,采用旋光度测定法测定青霉素钠。文献《青霉素皮试液含量测定两种方法的对照试验》(李程.首都医药.20007(2):),公开了用碘量法和紫外分光光度法进行含量测定。文献《考察青霉素皮试剂含量的稳定性》(袁波,张琳,袁玲.黑龙江医药.2003(1):28-29),公开了用高效液相色谱法对青霉素皮试剂中的青霉素G钠进行含量测定,其色谱条件为:色谱柱:Nova-pakC18柱(3.9mm×150nm);流动相:0.05mol/L磷酸二氢钾-乙腈(4:1);流速:1mL/min;检测波长:230nm;检测器扫描范围:200nm~250nm。该文献发现:皮试剂为冻干针剂时,因制备工艺的差异,导致制剂的稳定性差、杂质的种类与含量也比注射剂多,不利于检测。文献《高效液相色谱法测定青霉素皮试液的含量》(范义凤,季卫荣.医药导报.200423(12):952-953),公开了一种用高效液相色谱法测定青霉素皮试液的含量,其色谱条件为:色谱柱:Eclipse×OBC8柱(4.6mm×150nm);流动相:甲醇:水=40:60;流速:1.0mL/min;检测波长:240nm;进样量:20μL。该文献发现:医院自制的青霉素皮试液样品,按药典方法进行含量测定时效果不理想,主峰与杂质峰不能分离,仅在特定的流动相条件下才能改进分离度,较好的测定青霉素皮试液的含量。文献《不同厂家青霉素皮试剂质量的实验研究》(梁卫.广西中医学院学报.2000(4):61-63),公开了对不同厂家生产的青霉素皮试剂的含量测定,其色谱条件为:色谱柱:NPC8柱(4.6mm×180nm);流动相:乙腈:磷酸二氢钠=1:4;流速:1.2mL/min;检测波长:230nm;发现不同厂家生产的青霉素皮试剂质量差异较大,杂质含量颇多。由上可知,目前市售的、医院自制的青霉素皮试剂多为注射剂,少有冻干粉剂,且:①冻干粉剂尚无准确的质量检测标准,只能借鉴注射液的质量检测方法来改进;②冻干粉剂与注射剂相比,辅料不同,制备工艺也不同,冻干制剂稳定性控制的要求更高,杂质的种类也多,因此构建冻干粉剂的有关物质测定与含量测定的方法,技术难度更高;③不同厂家、不同制备工艺对于青霉素皮试冻干粉剂的质量影响较大,因此现有的质量检测方法通用性差、灵敏度低,不利于产品质控,大大增加了临床用药安全性的风险。综上所述,如何提供一种青霉素皮试冻干粉剂的质量检测方法,能够高效、灵敏的测定含量,提高该制剂的质量控制水平,是本领域技术人员急需解决的技术难题。技术实现要素:本发明目的在于提供了一种适用于青霉素皮试冻干粉剂的质量检测方法,通过改进药典方法,使得青霉素皮试冻干粉剂中的主峰、辅料峰和杂质峰能够清晰分离、样品含量能够精准测定,大大提高了皮试冻干粉剂的质量控制水平,解决了现有技术存在的上述缺陷。本发明所述的青霉素皮试冻干粉剂,系为国内某制药企业的独家产品,该处方由青霉素钠或其盐、赋形剂和注射用水组成,其中青霉素钠为≥160单位的固体青霉素钠,赋形剂由乳糖和甘露醇组成,重量比为乳糖:甘露醇=1:3。本发明所述的青霉素皮试冻干粉剂,制备工艺如下:取处方量的青霉素钠或其盐,加注射用水稀释,再加上述处方量的赋形剂,混合成药液,药液经微孔滤膜过滤除菌、分装后,冷冻干燥,密封,分装,即得。其中,冷冻干燥的工艺(即冻干工艺)具体包括如下步骤:(1)预冷:将分装后的样品放置于冷冻干燥机制品室板层,当样品温度降至-45℃时,压力为10~30Pa,保持3h;温度降至-55℃,压力为10~20Pa,保持2h;(2)升华:样品完全冻结后,开始抽真空,温度至-30℃,保持2h,当制品室板层温度升至-10℃,保持4h;再当温度升至0℃时,保持2h;(3)干燥:保持真空状态,温度升至40℃,保持4h,冻干结束;分装出品(4)分装:在10000级洁净区、局部100级洁净区,温度22℃,相对湿度45%,进行无菌分装,压塞,装量±3%;(5)贴签、装盒与装箱,制得待检产品。本发明提供的一种青霉素皮试冻干粉剂的质量检测方法,其技术方案具体如下:一种青霉素皮试冻干粉剂的质量检测方法,包括如下步骤:(1)对照品溶液的制备精密称取青霉素钠标准品0.02g,用乙腈溶解定容,混溶,配成浓度为0.2~2mg/ml的溶液,储存于棕色玻璃瓶中,并于2~8℃条件下放置保存;(2)供试品溶液的制备取青霉素皮试冻干粉剂0.2g,精密称定,适量置于量瓶中,加乙腈使细粉溶解,定容至2ml,摇匀,滤过,取续滤液作为供试品溶液;(3)高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,即可;色谱条件:固定相为:以十八烷基硅烷键合硅胶为填料的色谱柱;流动相为:甲醇、乙腈、水的混合液,检测波长为:225~240nm;流速为:0.5~1.5mL/min;柱温为:25~35℃;进样量为:20μl;等度洗脱。优选的,步骤(1)中,配成的对照品溶液浓度为2mg/ml。优选的,步骤(3)的色谱条件中,固定相为:Nova-pakC18的色谱柱。优选的,步骤(3)的色谱条件中,流动相为:甲醇、乙腈、水的体积比=(55~70):(5~20):25。更优选的,步骤(3)的色谱条件中,流动相各溶剂的体积比为:甲醇:乙腈:水=63:12:25。优选的,步骤(3)的色谱条件中,检测波长为:235nm。优选的,步骤(3)的色谱条件中,流速为:1.0mL/min。优选的,步骤(3)的色谱条件中,柱温为:30℃。优选的,步骤(3)中,供试品溶液青霉素钠主峰的面积不得超过对照品溶液峰面积,偏差不超过1.0%。本发明提供的青霉素钠皮试冻干粉剂质量检测方法,有益效有如下:(1)方法专属性强,专用于青霉素类药物皮试冻干粉剂的质量检测,可延长青霉素主峰在色谱检验系统中的保留时间,很好的消除辅料对检测的干扰;使得含量测定更准确;(2)方法重现性好,对不同批次的青霉素皮试冻干粉剂样品,按本方法进行重现性实验验证,重复进样6次,峰面积RSD为0.27%;(3)分离度高、检测结果精准,综合考虑了色谱柱、流动相的溶剂体系、检测波长、流速、柱温等因素变化对于检测结果的影响,缩短色谱检验系统平衡时间,提高色谱柱的耐用性,能很好地控制本发明药物的质量,避免有害物质未检出可能给患者临床用药安全带来的隐患;(4)样品来源的可获得性强,本发明提供的检测方法操作简单,通用性强,除青霉素钠皮试冻干粉剂外,其它青霉素类药物皮试冻干粉剂(如青霉素钾、苯唑青霉素钠、哌拉西林钠、磺苄西林钠、氨苄西林钠的皮试冻干粉剂),也可借鉴本方法。附图说明图1为在不同色谱柱条件下测定供试品含量的色谱图。图2为在不同流动相条件下测定供试品含量的色谱图。图1和图2中各序号含义:1.青霉素钠,2.乳糖,3.甘露醇。具体实施方式以下是本发明的具体实施例,对本发明的技术方案做进一步的描述,但是本发明的保护范围包括但并不限于这些实施例。凡是不背离本发明构思的改变或等同替代均包括在本发明的保护范围之内。实施例1:实验材料:仪器:Agilent1100液相色谱仪试剂:甲醇、乙腈(分析纯,500g/瓶,湖南师大化学试剂生产);超纯水,由实验室自制。色谱柱:Nova-pakC18柱(5μm,3.9mm×150nm,柱号0124977)实验方法:(1)精密称取青霉素钠标准品0.02g,用乙腈溶解定容,混溶,配成浓度为2mg/ml的溶液,储存于棕色玻璃瓶中,并于2~8℃条件下放置保存;(2)供试品溶液的制备取青霉素皮试冻干粉剂0.2g,精密称定,适量置于量瓶中,加乙腈使细粉溶解,定容至2ml,摇匀,滤过,取续滤液作为供试品溶液;(3)高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件为:固定相:Nova-pakC18柱,流动相:甲醇:乙腈:水=63:12:25,检测波长:235nm,流速:1.0ml/min,柱温:30℃,进样量:20μl,等度洗脱。(4)线性范围:取青霉素钠对照品分别钠溶液稀释至刻度,摇匀,分别进样,重复3次,按上述色谱条件测定峰面积(以吸收度A表示),浓度(C)对平均峰面积回归,得回归方程为A=2.563C+0.16452(r=0.9998),线性范围为120~600μg/ml。(5)精密度试验:取上述对照品溶液进样20μL,重复6次,测定青霉素主峰面积,测算所得峰面积RSD为0.2%和0.3%(n=6)。(6)重现性试验:取步骤(8)中同一批号样品(即批号分别为20150521、20150526、20150612的样品),以样品含量测定项下的方法,重复进样6次,峰面积RSD为0.27%。(7)稳定性试验:取浓度为0.1mg/ml对照品溶液,按对应色谱条件分别在第0、2、4、6、8、24小时进样测定,峰面积RSD分别为0.3%,0.2%,表明两种溶液在24小时内均稳定,且进样重现性良好。(8)样品含量测定:取青霉素皮试液样品直接进样20μL测定,以峰面积按回归方程计算,结果表明:批号为20150521、20150526、20150612的青霉素皮试液的含量分别为97.71%、99.33%、98.59%。实施例2色谱柱考察1.对照品溶液的制备精密称取青霉素钠标准品0.02g,用乙腈溶解定容,混溶,配成浓度为2mg/ml的溶液,储存于棕色玻璃瓶中,并于2~8℃条件下放置保存;2.供试品溶液的制备取青霉素皮试冻干粉剂0.2g,精密称定,适量置于量瓶中,加乙腈使细粉溶解,定容至2ml,摇匀,滤过,取续滤液作为供试品溶液;3.高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件:选择如下色谱柱进行比较:1#:NovapakC18柱(5μm,3.9mm×150nm);2#:EclipseOBC8柱(5μm,4.6mm×150nm);3#:AgilentTcC18柱(5μm,250×4.6mm),同时,流动相为特定体积比的甲醇-乙腈-水;检测波长为:235nm;柱温为:30℃;流速为:1.0mL/min;进样量为:20μl;等度洗脱。检测结果见表1和表2,不同色谱柱的色谱图结果见图1。表1不同色谱柱测定供试品含量结果的比较序号名称出峰时间(min)回收率(%)RSD(%)1#NovapakC18柱4.5899.81.12#EclipseOBC8柱3.4898.71.53#AgilentTcC18柱3.1595.60.9表2不同色谱柱测定供试品含量成分(%)Nova-pakC18柱EclipseOBC8柱AgilentTcC18柱对照品99.192.587.4青霉素钠96.494.380.9乳糖2.63.10.12甘露醇1.42.20.44结论:高效液相色谱法测定青霉素皮试剂冻干粉时,采用不同的色谱柱测定,Nova-pakC18柱4min左右出峰,测定时间5min左右,样品测定时间短;采用EclipseOBC8柱,7min左右出峰,测定时间10min左右,样品测定时间长;采用AgilentTc-C18柱,3min左右出峰,测定时间5min左右。但后两者(即2#和3#的色谱柱)不能较好的分离各个主成分峰、辅料峰和杂质峰,造成数据不准确。而采用Nova-pakC18柱,测定时间短,各峰分离效果好,效率高,适合大批量检验,值得推广使用。实施例3流动相考察1.对照品溶液的制备,方法参照实施例1。2.供试品溶液的制备,方法参照实施例1。3.高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件:选择如下流动相体系进行比较:1#:甲醇:水,其体积比分别为75:25、78:22、80:20;2#:甲醇:乙腈:水,其体积比分别为55:20:25、63:12:25、70:5:25;3#:甲醇:0.2%冰醋酸水溶液,其体积比分别为75:25、78:22、80:20;4#:乙腈:0.2%冰醋酸水溶液,其体积比分别为75:25、78:22、80:20;同时,色谱柱为:Nova-pakC18柱(5μm,3.9mm×150nm),检测波长为:235nm;柱温为:30℃;流速为:1.0mL/min;进样量为:20μl;等度洗脱。检测结果见表3,不同流动相体系的色谱图结果见图2。表3不同流动相体系中各峰的保留时间结论:①1#、3#和4#分别是不同比例的甲醇-水、甲醇-0.2%冰醋酸水溶液、乙腈-0.2%冰醋酸水溶液作为流动相体系,发现采用1#、3#或4#流动相体系,供试品的洗脱不明显,分离度差;而采用不同比例的甲醇-乙腈-水(即2#)作为流动相体系,对供试品的出峰顺序和保留时间都比1#、3#和4#体系好,各峰形集中,分离效果显著,且保留时间较长,符合检测要求,故将2#体系作为本发明的理想流动相。②通过标准偏差的比较,发现:当甲醇-乙腈-水的体积比为63:12:25时,检测效果最佳。实施例4流速考察1.对照品溶液的制备,方法参照实施例1。2.供试品溶液的制备,方法参照实施例1。3.高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件:色谱柱为:Nova-pakC18柱(5μm,3.9mm×150nm);流动相为特定体积比的甲醇-乙腈-水;检测波长为:235nm;柱温为:30℃;进样量为:20μl;等度洗脱,考察流速分别为0.5ml/min、1.0ml/min和1.5ml/min时对高效液相色谱检测的影响。结论:①流速分别为0.5ml/min、1.0ml/min和1.5ml/min时,检测结果差异不大,因此流速范围可选0.5~1.5ml/min;②当流速为0.5ml/min时,色谱峰略宽;当流速为1.5ml/min时,出峰略快,使得检测数据的采集略有偏差;而当流速为1.0ml/min时最佳,能够准确采集数据,因此最佳流速可选1.0ml/min。实施例5柱温的影响1.对照品溶液的制备,方法参照实施例1。2.供试品溶液的制备,方法参照实施例1。3.高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件:色谱柱为:Nova-pakC18柱(5μm,3.9mm×150nm);流动相为特定体积比的甲醇-乙腈-水;检测波长为:235nm;流速为:1.0ml/min;进样量为:20μl;等度洗脱,考察柱温分别为25℃、28℃、30℃和35℃时对高效液相色谱检测的影响。检测结果见表4。表4不同柱温下的色谱行为比较结论:①柱温分别为25℃、28℃、30℃和35℃时,含量测定结果的差异不大,因此柱温范围可选25~35℃;②当柱温为25℃、28℃时,发现柱压略高,主峰较宽,使得理论塔板数降低,所以分离度略低;而当柱温为30℃时,分离度最高,说明分离效果最佳,因此最佳柱温可选30℃。实施例6测定波长的选择1.对照品溶液的制备,方法参照实施例1。2.供试品溶液的制备,方法参照实施例1。3.高效液相色谱检测分别精密吸取对照品溶液、供试品溶液注入液相色谱仪,记录色谱图,记录时间为0~20min,供试品溶液中,青霉素钠主峰的面积不得超过对照品溶液峰面积(1.0%),即可;色谱条件:色谱柱为:Nova-pakC18柱(5μm,3.9mm×150nm);流动相为特定体积比的甲醇-乙腈-水;流速为:1.0ml/min;进样量为:20μl;等度洗脱,考察检测波长分别为225nm、235nm和240nm时对高效液相色谱检测的影响。对检测结果进行数据拟合后,得到如下的线性回归方程:S225=217.24ρ+137.08R2=0.9998;S235=327.87ρ+364.5R2=0.9998;S240=129.258ρ+145.7R2=0.9938;结论:于235nm波长处所建立的标准曲线方程的拟合度更高。检测结果显示,于225nm、235nm和240nm波长下测得供试品的主峰面积值分别为223.9、627.8和185.5,可见在225和240nm波长处的吸收较弱。所以,检测波长选择235nm最适宜,此时青霉素皮试冻干粉剂的检测灵敏度最高。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1