一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法与流程
本发明涉及医药领域,具体的说,本发明涉及一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法。
背景技术:
溶出度(dissolution):指活性药物从片剂、胶囊剂或颗粒剂等普通制剂在规定条件下溶出的速度和程度(即药物在溶出介质中达到某个时间点时释放的量),主要是考察药物在规定的时间点时的释放,能否达到标准中规定的限度。
溶出曲线(dissolutionprofile):是制剂在不同时间点获得的溶出度绘制的溶出度-时间曲线。溶出曲线反映了制剂的体外溶解和溶出过程,可通过比较仿制制剂与原研制剂体外多条溶出曲线相似性的方法,评价仿制药的质量。
从2012年起,国家药品监督管理局(nmpa)就启动了仿制药质量一致性评价工作,即将已经批准上市的仿制药,按与原研药品质量和疗效一致的原则,使仿制药在质量和疗效上与原研药一致。其中制定科学合理的固体制剂溶出曲线,是提高体内生物等效性试验成功率的重要步骤,并为将药品特征溶出曲线列入相应的质量标准提供依据,为药品批间质量的一致性、工艺变更前后药品质量的一致性提供保证。
目前,国内外药典中收载的各种混悬液,要求测定溶出度的较少,仅美国药典usp<42>中对醋酸甲地孕酮口服混悬液、非尔氨脂口服混悬液、吲哚美辛口服混悬液规定了溶出度测定要求(采用溶出度测定法第二法测定)。
当前市场上,第一/二法溶出仪主要为测定固体制剂的溶出度而设计,液体制剂用此仪器进行溶出度测定时,一般采用注射器手动加样的方式。混悬液溶出曲线对样品状态、加样位置、加样速度及加样针孔径等变化较敏感,从而影响平行检测样品溶出量结果的精密度。
当前市售醋酸甲地孕酮口服混悬液为
醋酸甲地孕酮混悬液市售制剂
市售制剂
技术实现要素:
本发明的目的在于克服现有药典资料的不足,提供一种能有效区分处方和工艺变量对醋酸甲地孕酮混悬剂体外释放溶出曲线的测定方法,并提高该方法的精密度和准确度,为药品批间质量的一致性提供保证。
为达上述目的,本发明提供了一种有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法,其中,所述方法包括如下步骤:
(1)配制溶出介质:包括将表面活性剂溶于水中配制成溶出介质的步骤,其中表面活性剂包括十二烷基硫酸钠(sds)或聚山梨酯80(吐温80),当表面活性剂为十二烷基硫酸钠时,十二烷基硫酸钠质量浓度范围为0.35%~0.45%,当表面活性剂为聚山梨酯80时,聚山梨酯80的质量浓度范围为0.8%-1.0%;
(2)待测样品制备:包括取待测的醋酸甲地孕酮口服混悬液充分摇匀,经减压脱气制得待测样品;
(3)溶出量测定:包括将待测样品经自动加样装置加入溶出介质中,按照溶出仪桨法进行测定,于不同取样时间点进行取样,测定不同时间点的溶出量。
根据本发明一些具体实施方案,其中,步骤(2)包括将待测的醋酸甲地孕酮混悬液剧烈振摇,以使得所述混悬液充分混匀,然后减压脱气30-60min,真空度维持在-50~-80kpa,得到待测样品。
所述剧烈振摇为本领域通常理解的剧烈振摇,而根据本发明一些具体实施方案,所述剧烈振摇为将样品瓶以每分钟至少80次的频率上下振摇。
根据本发明一些具体实施方案,其中,步骤(3)包括采用自动加样装置将待测样品加入溶出介质中,其中所述自动加样装置(如图9所示)至少由注射器2、注射泵1、加样针4、连接软管3(优选为硅胶材质连接软管)组成。
其中,所述注射泵可以为本领域常规的注射泵,而根据本发明一些具体实施方案,其中,步骤(3)注射泵包括单通道或多通道注射泵。
根据本发明一些具体实施方案,其中,步骤(3)包括加样量范围为0.5ml~10ml。
根据本发明一些具体实施方案,其中,步骤(3)包括加样位置为位于溶出介质液面下0.5-1.5cm处,优选为1cm。
根据本发明一些具体实施方案,其中,步骤(3)包括加样针为平头针,针头内径为0.5mm~1.54mm(对应的国际通用标准规格为21g~14g)。
根据本发明一些具体实施方案,其中,步骤(3)的加样时间为10-15s。
根据本发明一些具体实施方案,其中,步骤(3)取样时间点包括5min,10min,15min,20min,30min,45min,60min。
根据本发明一些具体实施方案,其中,步骤(3)包括溶出量测定方法包括高效液相色谱法或紫外分光光度法。
根据本发明一些具体实施方案,其中,步骤(3)所述高效液相色谱法的色谱条件包括:
检测器:紫外检测器;
色谱柱:以ods-3,4.6mm×150mm,5μm为色谱柱;
检测波长:为292nm;
柱温:35℃;
流动相:以乙腈-水(65:35)为流动相;
流速:1.2ml/min。
根据本发明一些具体实施方案,其中,步骤(3)所述紫外分光光度法检测波长为292nm。
根据本发明一些具体实施方案,其中,所述方法包括如下步骤:
(1)溶出介质制备:包括将表面活性剂十二烷基硫酸钠或聚山梨酯80溶于水中,其中十二烷基硫酸钠浓度范围为0.35%~0.45%,聚山梨酯80浓度范围为0.8%-1.0%;
(2)待测样品制备:包括取待测的醋酸甲地孕酮口服混悬液充分摇匀,经减压脱气30-60min制得待测样品;
(3)溶出量测定:将待测样品经自动加样装置加入溶出介质中,其中采用注射器量取5-20ml待测样品,以硅胶软管连接注射器和加样针,加样针头部置于溶出介质液面下0.5cm~1.5cm处,加样针内径约1.5mm,调节注射泵加样速度使加样时间控制在8~15s,溶出仪桨叶转速设置为30转/min,分别于5min,10min,15min,20min,30min,45min,60min取样,按照高效液相色谱法或紫外分光光度法测定相应的溶出量。
根据本发明一些具体实施方案,其中,步骤(1)还包括利用缓冲盐调节溶出介质的ph值的步骤,所述缓冲盐选自盐酸、磷酸盐、醋酸盐、冰醋酸、氢氧化钠和氢氧化钾中的一种或多种的混合,所述缓冲盐用量为将配制得到的溶出介质的ph值调节为1.0~7.0。
其中可以理解的是,步骤(1)在配制溶出介质时,可以加入缓冲盐,也可以不加入。
根据本发明一些具体实施方案,其中,步骤(1)包括将表面活性剂溶于水中,然后加入适当缓冲盐配制成溶出介质。
根据本发明一些具体实施方案,其中,步骤(1)包括将表面活性剂溶于水中配制溶出介质,以及将表面活性剂溶于水中后进一步加入缓冲盐配制溶出介质,以分别配制成缓冲盐加入量分别为0、以及将配制得到的溶出介质ph值调节为1.2、4.5和6.8溶出介质,所述缓冲盐选自盐酸、磷酸盐、醋酸盐、冰醋酸、氢氧化钠和氢氧化钾中的一种或多种的混合。
可以理解的是,步骤(1)所述溶出介质可以包括八种:
根据本发明一些具体实施方案,其中,十二烷基硫酸钠配制的溶出介质有四种,分别为:0.4%十二烷基硫酸钠水溶液(未加入缓冲盐);十二烷基硫酸钠质量浓度为0.4%、加入缓冲盐将配制得到的溶出介质ph调节为1.2;十二烷基硫酸钠质量浓度为0.4%、加入缓冲盐将配制得到的溶出介质ph调节为4.5;十二烷基硫酸钠质量浓度为0.4%、加入缓冲盐将配制得到的溶出介质ph调节为6.8;
根据本发明一些具体实施方案,其中,聚山梨酯80配制的溶出介质有四种,分别为:0.9%聚山梨酯80水溶液(未加入缓冲盐);聚山梨酯80质量浓度为0.9%、加入缓冲盐将配制得到的溶出介质ph调节为1.2;聚山梨酯80质量浓度为0.9%、加入缓冲盐将配制得到的溶出介质ph调节为4.5;聚山梨酯80质量浓度为0.9%、加入缓冲盐将配制得到的溶出介质ph调节为6.8。
根据本发明一些具体实施方案,其中,步骤(1)未加入缓冲盐、十二烷基硫酸钠质量浓度0.4%(或聚山梨酯80质量浓度0.9%)的溶出介质的配制过程为:称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于量瓶中,加700~800ml水使溶解,加水稀释至1000ml,混匀,脱气,即得。
根据本发明一些具体实施方案,其中,步骤(1)溶出介质ph值为1.2、十二烷基硫酸钠质量浓度0.4%(或聚山梨酯80质量浓度0.9%)的溶出介质的配制过程为:称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于量瓶中,加700~800ml水使溶解,加入7.65ml盐酸,加水稀释至1000ml,混匀,脱气,即得。
根据本发明一些具体实施方案,其中,步骤(1)溶出介质ph值为4.5、十二烷基硫酸钠质量浓度0.4%(或聚山梨酯80质量浓度0.9%)的溶出介质的配制过程为:称取2.99g醋酸钠置于量瓶中,加2mol/l醋酸溶液14ml,加水稀释至400ml;另称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于另一量瓶中,加500ml水使溶解,将两个量瓶中的溶液混合,用水稀释至1000ml,混匀,脱气,即得。
根据本发明一些具体实施方案,其中,步骤(1)溶出介质ph值为6.8、十二烷基硫酸钠质量浓度0.4%(或聚山梨酯80质量浓度0.9%)的溶出介质的配制过程为:称取6.0g磷酸二氢钠置于量瓶中,加0.2mol/l氢氧化钠溶液112ml,加水稀释至400ml;另称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于另一量瓶中,加500ml水使溶解,将两个量瓶中的溶液混合,用水稀释至1000ml,混匀,脱气,即得。
根据本发明一些具体实施方案,其中,步骤(3)还包括将所测得的溶出量绘制溶出曲线,以评估待测样品的体外溶出特性。
根据本发明一些具体实施方案,利用步骤(3)绘制的溶出曲线,采用相似因子f2对不同处方和工艺制剂与原研制剂的相似程度进行评估。
根据本发明一些具体实施方案,其中,待测样品的体外溶出曲线用于评估产品处方或工艺变化对醋酸甲地孕酮口服混悬液质量的影响。
根据本发明一些具体实施方案,其中,待测样品的体外溶出曲线用于预测产品体内生物等效性的差异。
根据本发明一些具体实施方案,其中,所述方法还包括配制对照品溶液的步骤,并在步骤(3)分别将对照品和待测样品经自动加样装置加入溶出介质中,按照溶出仪桨法分别进行测定,分别于不同取样时间点进行取样,测定不同时间点的溶出量。
根据本发明一些具体实施方案,其中,所述方法还包括:
(1)配制溶出介质:包括将表面活性剂溶于水中配制成溶出介质的步骤,其中表面活性剂包括十二烷基硫酸钠(sds)或聚山梨酯80(吐温80),当表面活性剂为十二烷基硫酸钠时,十二烷基硫酸钠质量浓度范围为0.35%~0.45%,当表面活性剂为聚山梨酯80时,聚山梨酯80的质量浓度范围为0.8%-1.0%;
(2)配制对照品溶液;
(3)待测样品制备:包括取待测的醋酸甲地孕酮口服混悬液充分摇匀,经减压脱气制得待测样品;
(4)溶出量测定:包括分别将对照品和待测样品经自动加样装置加入溶出介质中,按照溶出仪桨法分别进行测定,分别于不同取样时间点进行取样,测定不同时间点的溶出量。
综上所述,本发明提供了一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法。本发明的测定方法具有如下优点:
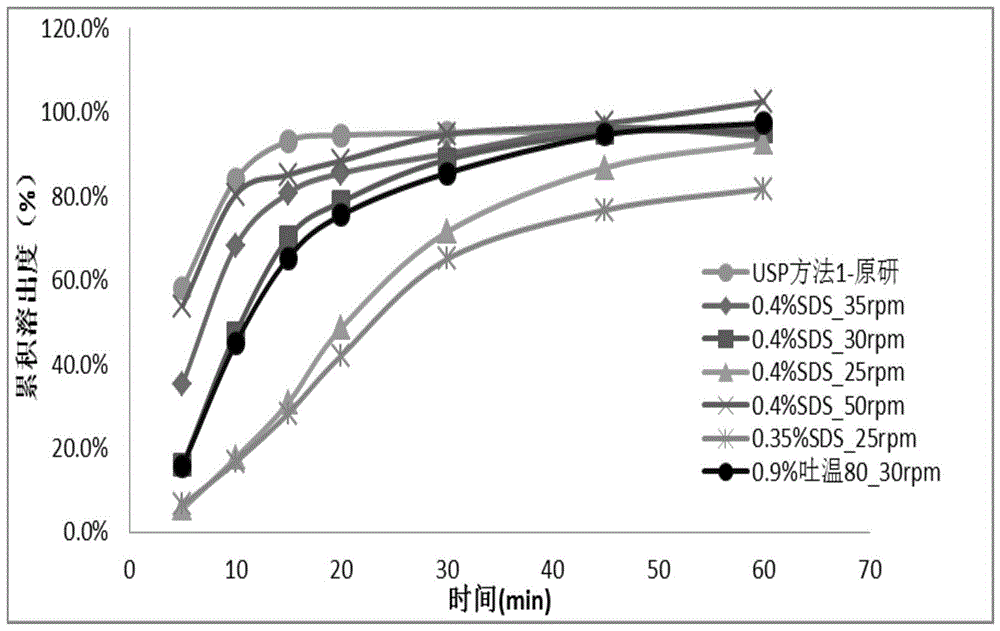
(1)本发明中,溶出介质设置为0.4%十二烷基硫酸钠溶液或0.9%聚山梨酯80溶液,转速设置为30转/分钟,通过大量实验表明:采用此溶出条件所测定的溶出曲线,较usp方法区分度更好,能够适度区分仿制药与原研药的质量优劣,同时能科学区分仿制药处方和工艺的差异,见图1。
(2)本发明中,规定待测样品制备方式为:剧烈振摇,使混悬剂充分混匀,减压脱气30-60min。混悬剂长期放置会出现沉降分层现象,如混和时间较短,不能确保瓶内不同位置的样品状态和含量均一致;剧烈振摇混合后,混悬剂内部会出现大量微小气泡,此时进行溶出曲线测定,相同取样时间点的溶出量会增加,溶出曲线区分度变差。大量实验表明,采用本发明待测样品制备方式,可在确保测试样品均一性的同时,提高溶出曲线测定方法的区分力。
(3)本发明中,溶出量测定样品加样方式采用自动加样装置,消除了不同人员,不同时间加样速度差异的干扰,显著提高了溶出曲线方法的精密度和准确度。
(4)本发明中,规定了加样位置、加样速度、加样用针头孔径,确保溶出曲线可重现。
(5)本发明方法所提供的混悬剂溶出曲线测定方法,具有科学,耐用,精密度高,重现性好等特点,对实现仿制药与原研药的质量一致性评价工作,药品批间质量的一致性、工艺变更前后药品质量的一致性提供可靠保证。
附图说明
图1为本发明实施例1中提供的原研制剂以usp方法1进行溶出曲线示意图,及溶出介质中sds浓度筛选和溶出仪转速筛选过程的原研制剂溶出曲线示意图;
图2为本发明实施例1中提供的原研制剂和自研制剂采用不同处理方式测定的溶出曲线示意图;
图3为本发明实施例1中提供的原研制剂采用不同加样时间测定的溶出曲线示意图;
图4为本发明实施例1中提供的原研制剂采用不同加样位置加样测定的溶出曲线示意图;
图5为本发明实施例1中提供的原研制剂采用不同孔径针头加样测定的溶出曲线示意图;
图6为本发明实施例1中提供的原研制剂采用自动加样与手动加样测定的溶出曲线示意图;
图7为采用实施例2中所提供一种具有区分力的测定醋酸甲地孕酮口服混悬液的溶出曲线的方法,测定出的原研制剂和3种不同处方和工艺制备的醋酸甲地孕酮口服混悬液的溶出曲线图;
图8为采用实施例2中所提供一种具有区分力的测定醋酸甲地孕酮口服混悬液的溶出曲线的方法,测定出原研制剂在4种不同ph溶出介质中的溶出曲线图;
图9为本发明采用的自动加样装置。
具体实施方式
以下通过具体实施例详细说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,不作为对本案可实施范围的限定。
实施例1
现有技术中,美国药典(usp<42>)中收载了测定醋酸甲地孕酮口服混悬液的溶出度方法,对原研制剂采用usp方法1进行溶出曲线测定:以0.5%的十二烷基硫酸钠(sds)水溶液为溶出介质,取900ml加入溶出杯中,设置溶出仪转速为25转/分钟,待溶出介质温度达到37±5℃时,使用注射器吸取4ml经混合后的醋酸甲地孕酮口服混悬液,于溶出介质表面注入溶出杯中,分别于5min,10min,15min,20min,30min,45min,60min时取溶液5ml,经0.45μm微孔滤膜过滤,取续滤液作为供试品溶液,以高效液相色谱法进行溶出量测定,色谱条件如下:
检测器:紫外检测器;
色谱柱:以ods-3,4.6mm×150mm,5μm为色谱柱;
检测波长:为292nm;
柱温:35℃;
流动相:以乙腈-水(65:35)为流动相;
流速:1.2ml/min。
结果表明使用usp方法1溶出结果过快(见图1),15分钟溶出量已达到85%,该溶出方法无区分力。
为了探索一个比较适合的方法,使醋酸甲地孕酮口服混悬液的溶出曲线达到较好的区分力,在进一步的研究试验中,分别考察了sds浓度变化、转速变化、样品处理方式变化、加样方式变化对醋酸甲地孕酮口服混悬液溶出曲线的影响。
通过大量实验表明,溶出介质sds的浓度为0.4%,转速为30转/分钟时,所测定的溶出曲线,较usp方法区分度更好,能够适度区分仿制药与原研药的质量优劣,同时能科学区分仿制药处方和工艺的差异,见图1。
混悬剂长期放置会出现沉降分层现象,如混合时间较短,不能确保瓶内不同位置的样品状态和含量均一致,从而影响溶出曲线方法的精密度;如剧烈振摇混合,混悬剂内部会出现大量微小气泡,此时进行溶出曲线测定,相同取样时间点的溶出量会增加,溶出曲线区分度变差。经实验表明,混悬剂经剧烈振摇混匀,再经减压脱气30-60min后,30min内相同取样点的溶出量变小,溶出曲线区分力更好,见表1、图2;
表1不同样品处理方式溶出量的差异
大量实验表明,混悬液加样时,加样速度、加样位置、加样针针头孔径等参数均会对溶出曲线造成影响,见图3-图6,本发明规定使用内径1.50mm的加样针,于溶出介质液面下0.5cm-1.5cm处进行加样,于10-15秒完成加样。
本发明中,溶出样品采用图9所示的实验室注射泵自动加样装置,消除了不同人员,不同时间加样速度差异的干扰,可保证始终匀速加样,控制第一和第二个点rsd均低于5%,显著提高了溶出曲线方法的精密度和准确度(如表2所示)。
表2不同加样方式溶出曲线及精密度结果
实施例2
本实施例中提供了一种测定醋酸甲地孕酮口服混悬液的溶出曲线的方法,包括如下步骤:
步骤(1)溶出介质的制备
溶出介质为0.4%十二烷基硫酸钠(或0.9%聚山梨酯80)水溶液,其配制过程为:称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于量瓶中,加700ml水使溶解,加水稀释至1000ml,混匀,脱气,即得。
溶出介质为ph1.2、含0.4%十二烷基硫酸钠(或0.9%聚山梨酯80)水溶液,其配制过程为:称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于量瓶中,加700~800ml水使溶解,加入7.65ml盐酸,加水稀释至1000ml,混匀,脱气,即得。
溶出介质为ph4.5、含0.4%十二烷基硫酸钠(或0.9%聚山梨酯80)水溶液,其配制过程为:称取2.99g醋酸钠置于量瓶中,加2mol/l醋酸溶液14ml,加水稀释至400ml;另称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于另一量瓶中,加500ml水使溶解,将两个量瓶中的溶液混合,用水稀释至1000ml,混匀,脱气,即得。
溶出介质为ph6.8、含0.4%十二烷基硫酸钠(或0.9%聚山梨酯80)水溶液,其配制过程为:称取6.0g磷酸二氢钠置于量瓶中,加0.2mol/l氢氧化钠溶液112ml,加水稀释至400ml;另称取4.0g十二烷基硫酸钠(或9.0g聚山梨酯80)置于另一量瓶中,加500ml水使溶解,将两个量瓶中的溶液混合,用水稀释至1000ml,混匀,脱气,即得。
在测定醋酸甲地孕酮口服混悬液的溶出曲线时,由于ph值的不同,需要测定醋酸甲地孕酮口服混悬液在每种ph溶出介质中的溶出曲线。
步骤(2)对照品溶液制备:
精确称取醋酸甲地孕酮对照品约18mg于100ml量瓶中,加入约2.5ml甲醇使溶解,用步骤1中溶出介质稀释定容至刻度,制成每1ml中约含180μg醋酸甲地孕酮的溶液,摇匀,作为对照品溶液。
步骤(3)待测样品制备
将待测的醋酸甲地孕酮混悬液剧烈振摇,以使得所述混悬液充分混匀,然后减压脱气30-60min得到待测样品。
步骤(4)供试品溶液制备:
取步骤(1)中的所述溶出介质,按照溶出度测定法第二法(桨法),设置转速为30转/分钟。使用一次性注射器吸取步骤(3)中经新鲜混合的混悬液(样品溶液)约20ml,置于实验室双通道注射泵(图9所示)上,使用硅胶连接软管连接注射器和加样针(内径1.5mm的针头),并使硅胶连接软管至取样针均冲满样品溶液(无气泡),将加样针位置固定于溶出介质液面下1.0cm处进行加样,加样时间为10秒,待溶出杯温度达到37±5℃,进行自动加样,计时,分别于5min,10min,15min,20min,30min,45min,60min时取溶液5ml,溶出液经0.45μm微孔滤膜过滤,取续滤液作为供试品溶液。
步骤(5)溶出量测定方法:
采用高效液相色谱法或紫外分光光度法进行溶出量测定。
高效液相色谱法的色谱条件为:
检测器:紫外检测器;
色谱柱:以ods-3,4.6mm×150mm,5μm为色谱柱;
检测波长:为292nm;
柱温:35℃;
流动相:以乙腈-水(65:35)为流动相;
流速:1.2ml/min。
紫外分光光度法的检测条件为:检测波长292nm。
步骤(6)溶出曲线测定
按照步骤(3)和步骤(4)制得供试品溶液,精密量取对照品溶液和供试品溶液10μl,注入液相色谱仪,并记录色谱图,按外标法以峰面积分别计算出原研制剂在5min,10min,15min,20min,30min,45min,60min时醋酸甲地孕酮的累计溶出量。重复以上步骤2次,计算原研制剂在5min,10min,15min,20min,30min,45min,60min时的平均累计溶出量,并绘制原研制剂在步骤(1)不同ph溶出介质中的溶出曲线。
步骤(7)评估:
采用相似因子法,利用原研制剂和仿制制剂在5min,10min,15min,20min,30min,45min,60min时的平均累计溶出量数据,计算相似因子f2,所述相似因子f2用于比较仿制制剂溶出曲线与原研制剂溶出曲线的相似性;利用相似因子f2的值判断相似性时,相似因子f2大于或等于50时,仿制制剂与原研制剂的溶出度曲线相似,相似因子f2小于50时,仿制制剂与原研制剂的溶出曲线不相似。
在进一步地优化方案中,可以优先选择10、15、20、30分钟四个时间点时,仿制制剂与原研制剂的溶出量数据,计算相似因子f2的值,不仅能达到相同效果,还可以简化计算过程,便于利用相似因子f2的值比较仿制制剂的溶出曲线与原研制剂的溶出曲线的相似性。
实施例3
按照实施例2中提供的一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法,分别对原研制剂、不同处方、不同工艺及最终处方工艺的醋酸甲地孕酮口服混悬液溶出曲线进行了测定,其中,
步骤(1)中,采用的溶出介质为在0.4%十二烷基硫酸钠水溶液;
步骤(5)中,具体采用高效液相色谱法进行溶出量测定;
通过测定,获得了原研制剂、不同处方制剂、不同工艺制剂及最终处方工艺制剂的溶出结果,见表3和图7。
表3本发明方法测定不同制剂溶出结果对比
由测定结果可见,本发明所提供的溶出曲线方法对不同处方、工艺的醋酸甲地孕酮口服混悬液有较好的区分力,可以有效地区分不同批次处方和工艺带来的差异。
实施例4
按照实施例2中提供的一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法,首先对原研制剂进行不同ph溶出曲线测定,其中,
步骤(1)中,采用的溶出介质分别为:实施例2中所述0.4%十二烷基硫酸钠水溶液,ph1.2、含0.4%十二烷基硫酸钠溶液,ph4.5、含0.4%十二烷基硫酸钠溶液,ph6.8、含0.4%十二烷基硫酸钠溶液;
步骤(5)中,具体采用高效液相色谱法进行溶出量测定;
通过测定,获得了原研制剂在四种溶出介质中的溶出曲线,如图8所示,可见原研制剂在四种溶出介质中的溶出曲线非常接近,利用本发明所提供的方法可将原研制剂的溶出曲线作为参考,对仿制制剂的相似性及质量一致性进行有效评估。
在进一步的实例中,按照实施例2中提供的一种具有区分力的醋酸甲地孕酮混悬液溶出曲线测定方法,对实施例3中最终处方工艺制剂进行不同ph溶出介质的溶出曲线进行了测定,通过相似因子f2对原研制剂和最终处方工艺制剂进行相了似性评估,结果见表4。
表4原研制剂与自研制剂相似性评估
其中,原研制剂为市售醋酸甲地孕酮口服混悬液为
自研制剂的处方与配比和原研制剂相同,制备过程包括:将醋酸甲地孕酮、聚乙二醇1450、聚山梨酯80、黄原胶、蔗糖、苯甲酸钠、枸橼酸(一水)、枸橼酸钠(二水)、柠檬香精等加水混合、经均质制得。
本发明进一步将实施例2-4的十二烷基硫酸钠替换为聚山梨酯80,并取得了与实施例2-4基本相同的效果。
综上所述,本发明溶出曲线方法不但具有很好的区分力,可用于评估产品处方或工艺变化对醋酸甲地孕酮口服混悬液质量的影响,以及预测产品体内生物等效性的差异,可为仿制制剂的质量控制和相似性评估提供体外依据。
- 还没有人留言评论。精彩留言会获得点赞!