一种阿托伐他汀钙中间体中共存杂质同时测定方法与流程
本发明涉及药物分析领域,更具体地,涉及一种同时测定m4中共存杂质含量的方法。
背景技术:
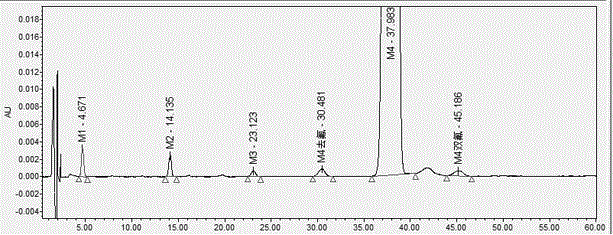
:阿托伐他汀钙(atv—ca)是由美国warner-lambert公司和pfizer公司于1997年共同开发上市的血脂调节药,可临床应用于治疗高胆固醇血症、抗动脉粥样硬化、降低血压、治疗糖尿病肾病,是市场上销售最好的3-羟基-3-甲基戊二酰辅酶a(hmg.coa)还原酯抑制剂。阿托伐他汀钙的合成路线有多种,其中中间体m4被广泛用于阿托伐他汀钙的合成,其分子式为c26h24fno3,化学名为(±)4-氟-α-[2-甲基-1-氧代丙基]-γ-氧代-n,β-二苯基苯丁酰胺,化学结构式如下:中间体m4的质量控制是阿托伐他汀钙有关物质和含量控制的前提。目前关于阿托伐他汀钙的质量控制的方法偏多,例如张晓峰等人(张晓峰,侯晓清,邓凤霞,等.hplc法测定阿托伐他汀钙有关物质[j].药物分析杂志,2014,034(008):1504-1508.)公开了hplc法测定阿托伐他汀钙有关物质的检测,对阿托伐他汀钙中可能存在的去氟阿托伐他汀(杂质a);阿托伐他汀对映体(杂质b);二氟阿托伐他汀(杂质c);阿托伐他汀内酯(杂质d);阿托伐他汀钙缩合物(杂质e)均进行了检测分析。然而,目前还没有文献报道关于阿托伐他汀钙中间体尤其是中间体m4的质量控制方法。因此,亟需提供一种阿托伐他汀钙中间体m4的质量控制方法,对于提高阿托伐他汀钙的质量,进而保证广大用药者的安全性具有重要意义。技术实现要素:本发明的目的是克服上述现有技术中的缺陷和不足,提供一种同时测定m4中共存杂质含量的方法。本发明上述目的通过以下技术方案实现:一种阿托伐他汀钙中间体中共存杂质同时测定方法,所述中间体为m4,所述共存杂质包括m1、m2、m3、m4双氟、m4去氟;所述方法采用高效液相色谱仪对样品进行检测,其中色谱检测条件包括:以体积比为29:12:59的乙腈-四氢呋喃-0.05mol/l乙酸铵作为流动相进行等度洗脱。中间体m4中主要有m1、m2、m3、m4双氟、m4去氟这五种难分离的杂质,其结构式分别如下表:多种杂质的存在会严重影响合成的阿托伐他汀钙的品质,从中间体m4中有效分离出以上五种难去除的杂质,对于提高中间体m4的质量,保证阿托伐他汀钙至关重要。然而从上表可知,m4双氟、m4去氟与中间体m4的结构极其相似,导致分离难度非常大。本申请的发明人通过大量的实验探索发现,建立hplc分离法,改变流动相及洗脱方式,能够将以上5种杂质与中间体m4有效分离,各已知杂质与未知杂质之间的分离度远大于1.0,且该方法具有强专属性,能有效地应用于中间体m4的质量控制。优选地,所述样品的配制方法如下:s1.配样:配制供试品溶液:取m4样品适量,加甲醇稀释制成1ml中约含1mg的溶液,摇匀,得到供试品溶液;配制对照溶液:精密量取供试品溶液1ml置50ml量瓶中,加甲醇稀释至刻度,摇匀后精密移取1ml置10ml量瓶中,加甲醇稀释至刻度,摇匀,得到对照溶液。优选地,所述色谱检测条件具体为:填充柱为辛烷基键合硅胶;色谱柱规格为250mm×4.6mm,粒径5μm;流速为1.0~1.7ml/min;柱温为35℃~40℃,溶剂为甲醇;进样量为20μl;检测波长为244nm。更优选地,所述的流速为1.5ml/min。更优选地,所述的柱温为35℃。与现有技术相比,本发明具有如下有益效果:本发明的阿托伐他汀钙中间体中共存杂质同时测定方法,能够将中间体m4与各杂质同时进行分离,从而有效控制中间体m4的质量。经化学药品分析方法验证,本方法简便、准确度高、线性、重复性良好,完全适用于阿托伐他汀钙中间体m4的质量控制,为保证阿托伐他汀钙的用药安全提供了理论支持。附图说明图1是实施例1中专属性混合溶液图谱。具体实施方式为了更清楚、完整的描述本发明的技术方案,以下通过具体实施例进一步详细说明本发明,应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明,可以在本发明权利限定的范围内进行各种改变。本发明所使用的仪器和原料均市售获得,具体信息如下:waterse2695-2998高效液相色谱仪,empower3控制系统;msa3.6p-oce-dm电子天平(赛多利斯科学仪器有限公司);pb-10酸度计(赛多利斯科学仪器有限公司);中间体m4对照品(批号为3-lxm-171-1),由trc提供;中间体m4(批号为2018029)由天地恒一制药股份有限公司提供;已知杂质m1对照品来源为trc-p335325;m2对照品来源为trc-b279785;m3对照品来源为毕得医药;m4去氟杂质对照品来源于ostresearchchemicals-u40-10055;m4双氟杂质对照品来源于ostresearchchemicals-u40-10056;乙腈来源于sigma;四氢呋喃来源于tedia;乙酸铵来源于广东光华;纯化水来源于杭州娃哈哈集团有限公司。实施例1一种阿托伐他汀钙中间体中共存杂质同时测定方法,其中中间体为m4,共存杂质包括m1、m2、m3、m4双氟、m4去氟;检测方法具体如下:s1.配样:配制供试品溶液:取m4样品适量,加甲醇适量,振摇使溶解,用甲醇稀释制成1ml中约含1mg的溶液,摇匀,得到供试品溶液;配制对照溶液:精密量取供试品溶液1ml置50ml量瓶中,加甲醇稀释至刻度,摇匀后精密移取1ml置10ml量瓶中,加甲醇稀释至刻度,摇匀,得到对照溶液;s2.检测:采用高效液相色谱仪对s1中的溶液样品进行检测,其中色谱检测包括以下条件:检测结果及分析:取中间体m4样品两批,按上述溶液制备方法配置及检测,记录至主峰保留时间的2倍,按自身对照法计算各有关物质的含量,结果见表1。表1批号20180292019032m1n.an.am2n.an.am3n.an.am4去氟0.02%m4双氟n.a其他杂质0.03%0.03%结果表明,主成分m4及各有关物质之间分离效果良好。(1)方法专属性试验分别配置溶剂、各杂质定位溶液及中间体m4加杂质的专属性混合溶液(1mg/mlm4+0.1%m1+0.1%m2+0.1%m3+0.1%m4去氟+0.1%m4双氟);分别精密量取上述溶液各20μl,依次注入液相色谱仪,记录色谱图,结果如附图1所示。实验结果表明:①溶剂不干扰中间体m4有关物质检测;②专属性混合溶液中各有关物质与定位溶液保留时间一致;③专属性混合溶液中各有关物质及主成分之间分离度良好。(2)线性与范围取中间体m4及其5种杂质适量,加溶剂溶解并稀释成50μg/ml的混合溶液作为线性贮备液(相当于对照浓度的5000%);依次稀释成相当于对照浓度的30%、50%、80%、100%、150%、200%溶液,分别进样20μl。以质量浓度c(μg/ml)为横坐标,各组分峰面积a为纵坐标,进行线性回归,各组分回归方程见表2。表2组分名称浓度范围(μg/ml)线性方程相关系数响应因子m10.3302~2.2016a=46365c+1413.40.99903%m20.3124~2.0824a=44933c+2478.50.99836%m30.2996~1.9976a=21691c+1923.30.99647%m4去氟0.3080~2.0536a=33945c+2390.00.99666%m40.3102~2.0680a=41385c+3667.60.99937%m4双氟0.3127~2.0848a=39110c+1403.40.99764%各组分的相关线相关系数均大于0.99,且响应因子小于10%,本发明检测方法的线性关系良好。(3)精密度试验取线性与范围试验中的100%溶液,按“2.1项”测定方法,精密量取20μl,注入液相色谱仪,连续进样6针。以峰面积计算,6个组分的rsd分别为1.2%、1.9%、1.1%、1.5%、0.8%、1.1%,均小于3%。说明本发明检测方法的精密度良好。(4)检测限与定量限以信噪比s/n≈3为检测限(lod),信噪比比s/n≈10为定量限(loq),中间体m4及其5种杂质的loq和lod见表3。表3组分名称lod/ngloq/ngm12.20136.6040m22.08276.2480m31.99735.9920m4去氟2.05336.1600m42.06806.2040m4双氟2.08476.2540各组分的lod和loq均小于10%。(5)准确度取中间体m4样品10份每份10mg,精密加入各杂质混合溶液(50%、100%、150%各三份,不加样1份),分别指10ml量瓶中,加溶剂溶解并稀释至刻度,作为回收率供试品溶液,另制备100%杂质混合对照溶液2份,按外标法进行回收率测定,结果见表4。表4组分名称平均回收率rsdm1102.5%4%m2101.7%6%m396.7%5%m4去氟94.2%从表3中可以看出,各杂质的平均回收率均在90%~110%之间,说明本发明的方法准确度良好。对比例1一种阿托伐他汀钙中间体中共存杂质同时测定方法,其中中间体为m4,共存杂质包括m1、m2、m3、m4双氟、m4去氟;其中检测方法与实施例1基本相同,其区别在于,色谱检测条件不同,具体如表5。表5其中梯度洗脱程序如表6。表6检测结果:无法检出m4双氟、m4去氟的出峰。m4与各杂质,尤其是m4双氟、m4去氟不能有效分离。以上实施例使用具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些不偏离本发明主旨的修改或改进,这对本领域技术人员而言是显而易见的。因此,这些不偏离本发明主旨基础上所做的修改或改进,均属于本发明要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1