用高效液相色谱法分离盐酸米托蒽醌中杂质的方法与流程
1.本发明属于药物分析技术领域,具体涉及用高效液相色谱法分离盐酸米托蒽醌中杂质的方法。
背景技术:
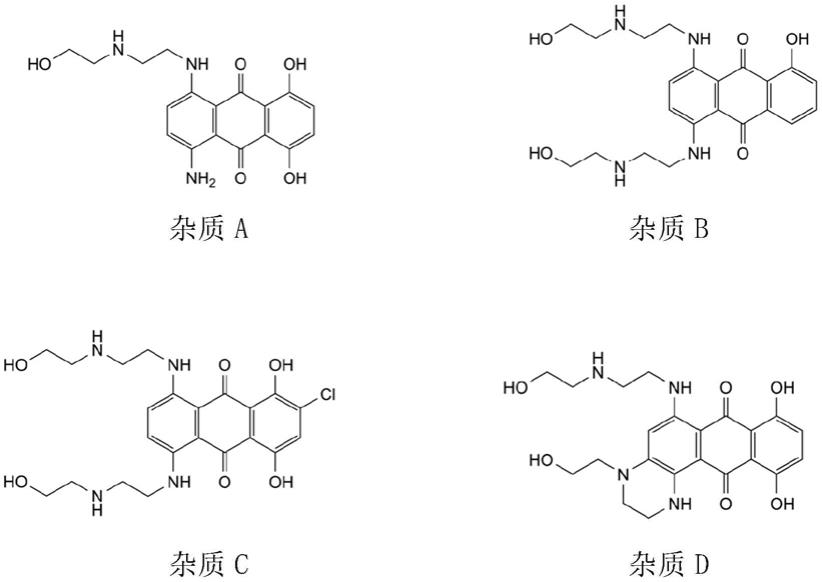
2.米托蒽醌是一种化学合成的广谱抗生素,在其衍生物(例如盐酸米托蒽醌)具体制备的过程中,不可避免引入各种杂质,如工艺杂质、降解杂质等,而这些杂质不容易检测和去除。为解决此问题,本发明人对米托蒽醌(尤其是盐酸米托蒽醌)的合成工艺和强制降解做了大量研究,结果发现在所制备的盐酸米托蒽醌中可能存在的有机杂质包括原材料、中间体、副产物等9种之多,例如杂质a、杂质b、杂质c、杂质d、lmtx、elr、tha、ltha、atha等,其中杂质a、杂质b、杂质c、杂质d、杂质lmtx、杂质elr、杂质ltha、杂质atha、杂质tha结构式如下:
3.[0004][0005]
盐酸米托蒽醌的制备和存储过程中可能会产生多种杂质,而产品中杂质的种类和含量直接影响药品质量和用药安全性,因此在使用之前需要对盐酸米托蒽醌中相关的杂质含量进行测定,以保证盐酸米托蒽醌的质量可控。
[0006]
目前中国药典2020年版二部公开了一种分离盐酸米托蒽醌中有关物质含量的方法。该方法以十八烷基硅烷键合硅胶为填充剂,以0.03mol/l庚烷磺酸钠溶液
‑
乙腈(70:30)为流动相,分离盐酸米托蒽醌中单个杂质含量。
[0007]
美国药典43版公开了一种分离盐酸米托蒽醌中有关物质含量的方法,该方法使用苯基柱,以水
‑
乙腈
‑
0.43mol/l庚烷磺酸钠溶液(750:20:25)为流动相,分离盐酸米托蒽醌中单个杂质含量。
[0008]
上述公开的方法采用等度洗脱操作简便,但当杂质种类较多时,难以完全分离,采用c18柱或苯基硅烷键合硅胶柱和等度洗脱方式,虽然方法操作简单,对仪器要求较低,对简单杂质体系分离度较好,但对胺类化合物的保留不强,对复杂杂质体系的分离效果较差,且分析时间较长,后出峰的组分色谱峰越宽,导致容易拖尾,分离效果变差。
技术实现要素:
[0009]
本发明可以解决的技术问题是:用高效液相色谱法分离盐酸米托蒽醌中的多种杂
质,实现对盐酸米托蒽醌的准确监控和质量的控制。
[0010]
有鉴于此,本发明的目的是提供一种用高效液相色谱法分离盐酸米托蒽醌中杂质的方法。
[0011]
本发明所述盐酸米托蒽醌的结构式如(
※
)所示:
[0012][0013]
为实现本发明的目的,本发明采用以下技术方案:
[0014]
用高效液相色谱法分离盐酸米托蒽醌中杂质的方法,所述高效液相色谱法分析过程中采用的色谱柱是以苯基
‑
己基键合硅胶为填料,以流动相ⅰ和流动相ⅱ按体积比例混合为流动相进行线性梯度洗脱,其中所述流动相ⅰ为醋酸铵缓冲液,所述流动相ⅱ为乙腈,所述梯度比例为:
[0015]
0min,流动相ⅰ与流动相ⅱ的体积比为85:15;
[0016]
14min,流动相ⅰ与流动相ⅱ的体积比为68:32;
[0017]
31min,流动相ⅰ与流动相ⅱ的体积比为62:38;
[0018]
36min,流动相ⅰ与流动相ⅱ的体积比为30:70;
[0019]
36.1min,流动相ⅰ与流动相ⅱ的体积比为5:95;
[0020]
37min,流动相ⅰ与流动相ⅱ的体积比为5:95;
[0021]
37.1min,流动相ⅰ与流动相ⅱ的体积比为85:15;
[0022]
42min,流动相ⅰ与流动相ⅱ的体积比为85:15。
[0023]
优选地,所述色谱法分析的色谱条件包括:
[0024]
色谱柱:柱长150mm,填料粒径5μm,内径4.6mm;
[0025]
检测器:光电二极管阵列检测器;
[0026]
检测波长:盐酸米托蒽醌、杂质a、杂质b、杂质c、杂质d和lmtx均为254nm,ltha为473nm、tha为516nm、atha为557nm、elr为611nm;
[0027]
柱温:35℃;
[0028]
流速:2.0ml/min。
[0029]
优选地,所述醋酸铵缓冲液的制备方法包括:将醋酸铵固体溶解于水中得溶液,向所述溶液中加入三氟乙酸调节所述溶液的ph。
[0030]
优选地,所述ph为2.0。在本发明中,用三氟乙酸调节所述溶液的ph使最终得到的醋酸铵缓冲液的ph为2.0。
[0031]
优选地,所述醋酸铵缓冲液中醋酸铵的浓度质量分数为0.10%。
[0032]
优选地,所述方法至少能分离出盐酸米托蒽醌中存在的以下九种杂质:杂质a、杂质b、杂质c、杂质d、杂质lmtx、杂质elr、杂质ltha、杂质atha和杂质tha。
[0033]
光电二极管阵列检测器是一种光学多通道检测器,在本发明中,采用光电二极管阵列检测器可同时检测盐酸米托蒽醌样品在所有波长的吸收,光电二极管阵列检测器具有灵敏度高、噪音低、线性范围宽、对流速和温度的波动不敏感等优点。
[0034]
本发明采用的色谱柱为苯基己基键合硅胶为填充剂的色谱柱(如luna phenyl
‑
hexyl色谱柱)进行检测,该色谱柱采用苯基
‑
己基键合相,可增加芳香和极性/胺类化合物的保留时间,有利于减少键合相的水解,使芳香和极性/胺类化合物具有重现性好、能够稳定存在的优势。
[0035]
在本发明中,必要时需要使用过滤器,使用过滤器的目的是过滤样品和各流动相中的细小颗粒,使样品和各流动相中没有颗粒成分,以保证色谱检测的顺利进行,在本发明中使用0.22μm过滤器或0.45μm过滤器。
[0036]
本发明的有益效果:
[0037]
(1)采用苯基己基色谱柱,增加盐酸米托蒽醌中杂质中的芳香和极性/胺类化合物的保留时间,有利于减少键合相的水解,使芳香和极性/胺类化合物具有重现性好、能够稳定存在的优势。
[0038]
(2)使用线性梯度洗脱程序,可以保证盐酸米托蒽醌中各杂质之间的相互分离,从而提高分离效果。
[0039]
(3)采用光电二极管阵列检测器可同时检出盐酸米托蒽醌中的不同波长的杂质,可以分离出复杂的杂质体系,专属性好,为盐酸米托蒽醌的质量控制提供了有效保障。
[0040]
(4)本发明方法能够有效分离盐酸米托蒽醌中的相关杂质,可以作为盐酸米托蒽醌质量控制的重要组成部分。
附图说明
[0041]
图1:实施例一的第一系统适用性溶液的hplc图谱;
[0042]
图2:实施例二的第二系统适用性溶液的hplc图谱;
[0043]
图3:杂质a、b、c、d、lmtx的线性关系(峰面积
‑
浓度)图;
[0044]
图4:杂质elr的线性关系(峰面积
‑
浓度)图;
[0045]
图5:杂质ltha的线性关系(峰面积
‑
浓度)图;
[0046]
图6:杂质atha的线性关系(峰面积
‑
浓度)图;
[0047]
图7:杂质tha的线性关系(峰面积
‑
浓度)图。
具体实施方式
[0048]
通过下面给出的本发明的具体实施例可以进一步清楚地了解本发明,但它们不是对本发明的限定。
[0049]
在本发明的说明书附图图1中的米托蒽醌为本发明的盐酸米托蒽醌。
[0050]
实施例一
[0051]
分离盐酸米托蒽醌中的杂质a、杂质b、杂质c、杂质d、杂质lmtx
[0052]
第一步:流动相的配制:
[0053]
流动相ⅰ:将1g醋酸铵固体溶解于1l水中得溶液,向溶液中加入三氟乙酸调节溶液的ph至2.0并用0.45μm过滤器过滤,得到0.10%醋酸铵缓冲液;
[0054]
流动相ⅱ:由乙腈配制;
[0055]
在本实施例一中,用流动相ⅰ和流动相ⅱ按流动相ⅰ:流动相ⅱ为850:150的体积比混合配制得稀释剂;
[0056]
第二步:第一供试品溶液的制备:
[0057]
精密称取20mg盐酸米托蒽醌样品加入50ml量瓶中,并向50ml量瓶中加入稀释剂使50ml量瓶中的盐酸米托蒽醌样品溶解,再向50ml量瓶中加入稀释剂稀释至50ml量瓶的刻度并混合均匀,得第一供试品溶液;
[0058]
第三步:对照溶液的制备:
[0059]
精密量取第一供试品溶液1ml,置100ml量瓶中,用稀释剂稀释至100ml,得对照溶液。
[0060]
第四步:第一系统适用性溶液的制备:
[0061]
精密称取杂质a、杂质b、杂质d和杂质lmtx的对照品各2mg和盐酸米托蒽醌峰鉴别对照品(含杂质c)5mg,加入10ml量瓶中,并向10ml量瓶中加入适量稀释剂超声5分钟使样品溶解,再用稀释剂稀释至10ml并摇匀,必要时经0.45μm过滤器过滤,得第一系统适用性溶液;
[0062]
第五步:第一系统适用性溶液验证
[0063]
吸取5μl的第一系统适用性溶液注入高效液相色谱仪中进行检测,其中色谱条件包括:
[0064]
色谱柱:luna phenyl
‑
hexyl色谱柱,柱长150mm,内径4.6mm,填料粒径5μm;
[0065]
流动相:流动相ⅰ:0.10%醋酸铵缓冲液、流动相ⅱ:乙腈;
[0066]
检测器:光电二极管阵列检测器;
[0067]
检测波长:254nm;
[0068]
柱温:35℃;
[0069]
流速:2.0ml/min;
[0070]
检测分析时间:42min;
[0071]
采用上述色谱条件进行线性梯度洗脱,其中线性梯度洗脱程序见表1:
[0072]
表1:线性梯度洗脱程序
[0073]
[0074]
线性梯度洗脱完成后进入光电二极管阵列检测器检测,记录色谱图,结果见图1。
[0075]
检测出盐酸米托蒽醌、杂质a、杂质b、杂质c、杂质d的波长为254nm。
[0076]
第六步第一供试品溶液中杂质的检测:
[0077]
精密量取第一供试品溶液与对照溶液各5μl,分别注入液相色谱仪,记录色谱图。
[0078]
杂质的计算:
[0079]
第一供试品溶液色谱图中若出现杂质a、杂质b、杂质c、杂质d、杂质lmtx峰(相对保留时间分别为杂质lmtx 0.6、杂质b 0.9、杂质c1.6、杂质a1.9、杂质d 2.1),按照主成分自身对照法计算各杂质含量,如下:
[0080][0081]
p:第一供试品溶液中杂质的百分含量;
[0082]
x:杂质a、杂质b、杂质c、杂质d、杂质lmtx中的任意一种。
[0083]
由色谱图图1分析可知,第一系统适用性溶液的出峰顺序依次为杂质lmtx、杂质b、盐酸米托蒽醌、杂质c、杂质a和杂质d,由表2可知,杂质lmtx、杂质b、盐酸米托蒽醌、杂质c、杂质a和杂质d峰之间的分离度均不低于1.5,符合《中国药典》中的规定,因此,可说明杂质a、杂质b、杂质c、杂质d和杂质lmtx的分离效果良好。
[0084]
表2第一系统适用性溶液中各杂质的保留时间及分离度
[0085][0086]
实施例二
[0087]
分离盐酸米托蒽醌中的杂质elr、杂质ltha、杂质atha、杂质tha
[0088]
第一步:第二供试品溶液的制备:
[0089]
精密称取100mg盐酸米托蒽醌样品加入50ml量瓶中,并向50ml量瓶中加入适量四氢呋喃,超声5min使盐酸米托蒽醌样品溶解,再向超声后的50ml量瓶中加入四氢呋喃至50ml量瓶的刻度并混合均匀,经0.22μm过滤器过滤,得第二供试品溶液;
[0090]
第二步:混合对照品溶液的制备:
[0091]
精密称取杂质elr、杂质ltha、杂质atha、杂质tha的对照品各2mg并分别加入到四个不同的100ml量瓶中,向各100ml量瓶中加入适量四氢呋喃,分别超声5min使各100ml量瓶内的杂质对照品溶解,再分别向各100ml量瓶中加入四氢呋喃至各100ml量瓶的刻度并混合
均匀,分别得到elr、ltha、atha和tha的对照品贮备液,再精密量取10ml上述各对照品贮备液放置在新的100ml量瓶中,并向新的100ml量瓶中加入四氢呋喃至新的100ml量瓶的刻度并混合均匀,得到混合对照品溶液;
[0092]
第三步:第二系统适用性溶液验证
[0093]
吸取15μl混合对照品溶液注入高效液相色谱仪中进行检测,采用与实施例一相同的色谱条件进行线性梯度洗脱,记录色谱图,结果见图2。
[0094]
第四步 第二供试品溶液中杂质的检测
[0095]
精密量取第二供试品溶液与混合对照品溶液各15μl,分别注入液相色谱仪,采用与实施例一相同的色谱条件进行检测,记录色谱图。检测波长分别为盐酸米托蒽醌254nm、ltha 473nm、tha 516nm、atha 557nm和elr 611nm。
[0096]
杂质的计算:
[0097]
第二供试品溶液色谱图中若出现杂质elr、杂质ltha、杂质atha、杂质tha峰,按照外标法计算各杂质组分含量,如下:
[0098][0099]
w:第二供试品溶液中的杂质含量。
[0100]
由色谱图图2分析可知,第二系统适用性溶液的出峰顺序依次为杂质elr、杂质ltha、杂质atha、杂质tha,由表3可知,杂质elr、杂质ltha、杂质atha、杂质tha峰之间的分离度均不低于1.5,符合《中国药典》中的规定,因此,可说明杂质elr、杂质ltha、杂质atha、杂质tha的分离效果良好。
[0101]
表3第二系统适用性溶液中各杂质的保留时间及分离度
[0102][0103]
实施例三
[0104]
盐酸米托蒽醌中各杂质的线性验证
[0105]
分别配制盐酸米托蒽醌中各杂质的线性系列溶液,采用与实施例一相同的色谱条件进行检测,进行线性验证研究。由于在实施例一中杂质a、杂质b、杂质c、杂质d、杂质lmtx使用的是主成分自身对照法,即是以盐酸米托蒽醌为主成分自身对照法分离出的,故在本实施例三中杂质a、杂质b、杂质c、杂质d、杂质lmtx的线性溶液取盐酸米托蒽醌对照品进行线性研究,另外,在本实施例三中各杂质的线性水平
‑
1、线性水平
‑
3、线性水平
‑
5溶液分别配制3份。
[0106]
①
杂质a、杂质b、杂质c、杂质d、杂质lmtx的线性结果见表4和图3:
[0107]
表4杂质a、b、c、d、lmtx的线性结果
[0108][0109]
表4所示的结果在图3线性关系(峰面积
‑
浓度)图)中可以直观的显示,其中盐酸米托蒽醌在0.00040~0.0056mg/ml,即在盐酸米托蒽醌中含量限度的0.1~1.4%浓度范围内,线性方程为y=5551447.90x
‑
285.03,r2=0.999,符合r2≥0.99的要求,线性关系良好。
[0110]
②
杂质elr的线性结果见表5和图4:
[0111]
表5杂质elr的线性结果
[0112][0113]
表5所示的结果在图4(elr线性关系(峰面积
‑
浓度)图)中可以直观的显示,其中elr在0.0010~0.0028mg/ml,即在盐酸米托蒽醌中含量限度的0.05~0.14%浓度范围内,线性方程为y=31616127.79x
‑
1881.74,r2=0.999,符合r2≥0.99的要求,线性关系良好。
[0114]
③
杂质ltha的线性结果见表6和图5:
[0115]
表6杂质ltha的线性结果
[0116]
[0117][0118]
表6所示的结果在图5(ltha线性关系(峰面积
‑
浓度)图)中可以直观的显示,其中ltha在0.0010~0.0028mg/ml,即在盐酸米托蒽醌中含量限度的0.05~0.14%浓度范围内,线性方程为y=13716895.95x
‑
8222.23,r2=0.998,符合r2≥0.99的要求,线性关系良好。
[0119]
④
杂质atha的线性结果见表7和图6:
[0120]
表7杂质atha的线性结果
[0121]
[0122][0123]
表7所示的结果在图6(atha线性关系(峰面积
‑
浓度)图)中可以直观的显示出,其中atha在0.0010~0.0028mg/ml,即在盐酸米托蒽醌中含量限度的0.05~0.14%浓度范围内,线性方程为y=22962974.86x
‑
133.97,r2=0.999,符合r2≥0.99的要求,线性关系良好。
[0124]
⑤
杂质tha的线性结果见表8和图7:
[0125]
表8杂质tha的线性结果
[0126][0127][0128]
表8所示的结果在图7(tha线性关系(峰面积
‑
浓度)图)中可以直观的显示,其中tha在0.0010~0.0028mg/ml,即在盐酸米托蒽醌中含量限度的0.05~0.14%浓度范围内,线性方程为y=14523819.83x
‑
279.61,r2=0.999,符合r2≥0.99的要求,线性关系良好。
[0129]
综上所述,杂质a、b、c、d、lmtx和杂质elr、杂质ltha、杂质atha、杂质tha的线性验
证结果表明上述各杂质的浓度均在其设计的理论浓度范围内,同时满足线性验证结果与试样中被测物质浓度直接呈正比关系要求,且线性关系良好。
[0130]
实施例四
[0131]
溶液稳定性的评价
[0132]
本实施例四的稀释剂与实施例一中的稀释剂相同。
[0133]
①
盐酸米托蒽醌溶液稳定性评价
[0134]
通过在3天内测定浓度水平为100%试验浓度的盐酸米托蒽醌的含量变化,以此评价在稀释剂中的盐酸米托蒽醌溶液稳定性。
[0135]
称取19.95mg盐酸米托蒽醌加入50ml量瓶中并加入稀释剂至50ml量瓶的刻度,混合均匀,得盐酸米托蒽醌溶液,并将盐酸米托蒽醌溶液保存在5~8℃的冰箱中,按相同的方法再配制2瓶与上述规格相同的盐酸米托蒽醌溶液并一同保存在5~8℃的冰箱中,采用与实施例一相同的色谱条件每天取1瓶盐酸米托蒽醌溶液进行检测,检测结果见表9:
[0136]
表9盐酸米托蒽醌溶液的稳定性评价
[0137]
时间盐酸米托蒽醌峰面积差值%第0天*23763470.0第1天23779440.1第2天23907960.6
[0138]
注:*稳定性溶液配制当天。
[0139]
评价标准:第n天与第0天的盐酸米托蒽醌峰面积的差值不得过
±
2%,其中n为1或2。
[0140]
结论:由表9的实验结果可知,盐酸米托蒽醌溶液可保持稳定2天。
[0141]
②
杂质elr、ltha、atha、tha的溶液稳定性评价
[0142]
通过在7天内测定相对于盐酸米托蒽醌的含量,浓度为10%的elr、ltha、atha和tha的溶液中elr、ltha、atha、tha的含量变化,以此评价在稀释剂中的elr、ltha、atha、tha溶液稳定性。
[0143]
称取20.29mg elr、20.31mg lhta、29.79mg atha和21.01mg tha的对照品分别放入四个50ml量瓶中,向各50ml量瓶加入稀释剂至各50ml量瓶的刻度,混合均匀,分别得到elr、ltha、atha、tha溶液并将各溶液保存在
‑
18℃的冰箱中,按相同的方法再配制6瓶与上述规格相同的各溶液并一同保存在
‑
18℃的冰箱中,采用与实施例一相同的色谱条件每天各取1瓶elr、ltha、atha和tha溶液进行检测,检测结果见表10:
[0144]
表10杂质elr、ltha、atha、tha溶液的稳定性评价
[0145][0146]
注:*稳定性溶液配制当天。
[0147]
评价标准:第n'天与第0天的各杂质峰面积的差值不得过
±
2%,其中n'为1、2、3、4、5或6。
[0148]
结论:由表10的实验结果可知,elr溶液可保持稳定6天;ltha溶液可保持稳定0天,ltha溶液应临用现制;atha溶液可保持稳定6天;杂质tha溶液可保持稳定6天。
[0149]
以上所述仅是本发明的实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1