白酒或白酒发酵过程样本中氰化物含量的检测方法与流程
1.本发明涉及技术检测领域,特别是涉及一种白酒或白酒发酵过程样本中氰化物含量的检测方法。
背景技术:
2.氰化物为剧毒物质,中毒者,轻则流涎、呕吐、腹泻,重则呼吸困难,全身抽搐,昏迷,在短时间内死亡。因此,食品安全国家标准gb2757-2012要求:蒸馏酒及其配制酒中氰化物含量不得超过8.0mg/l。
3.白酒中的氰化物主要来源于原料,一般而言,以谷物,如高粱、小麦、大麦、玉米为原料酿造的酒,由于这些原料中植物氰苷含量极微,酿造过程中氰苷经过水解可产生的氰化物也少,因此酿造出来的酒中的氰化物含量也极少。而以木薯、野生植物根茎酿造的酒,其氰化物的含量较高。这是因为木薯是生产酒精的良好原料,生产酒精用的木薯是块根部分,其外表皮含氢氰酸,所含的氰苷含量也较多,因此,用木薯及野生植物根茎酿造的酒中氰化物含量较高。若白酒配料表中有木薯或代用品的,那么基本都会有氰化物检出,检测值在国家标准限量范围内属于安全食品,但若生产工艺不当,造成氰化物含量超标,就容易造成白酒的安全隐患。白酒中氰化物含量超标与白酒的酿造工艺也有很大关系,如生产者用木薯、野生植物根茎直接酿酒,生产过程中又没有很好地对原料进行处理,在酿酒过程中会产生氢氰酸,代入白酒中,导致氰化物含量超标。另外,还有一些是用了氰化物含量超标的木薯食用酒精造成的。
4.传统检测白酒中氰化物的方法为化学分析法,如滴定法、试纸条快速检测法,其方法简便、快速,对仪器设备要求低,但存在局限性,定性尚可,定量结果不够准确。
5.传统检测食品中氰化物含量的方法较多,如采用顶空-气相色谱法测定水中氰化物的含量,但是白酒与水不同,白酒中高浓度的乙醇以及低沸点的基质均会对检测过程产生影响,从而影响检测结果的准确性、精密度与检测限。
技术实现要素:
6.基于此,本发明提供了一种白酒或白酒发酵过程样本中氰化物含量的检测方法,所述检测方法获得的检测结果准确性、精密度高,检出限与定量限较低。
7.本发明通过如下技术方案实现。
8.一种白酒或白酒发酵过程样本中氰化物含量的检测方法,包括如下步骤:
9.将含氰离子的标准品与第一碱性溶液混合,然后与第一酸性溶液混合,再与第一氯胺t溶液混合,制备标准品溶液;
10.将待测样品与第二碱性溶液混合,然后与第二酸性溶液混合,再与第二氯胺t溶液混合,制备试样溶液;
11.将所述标准品溶液与所述试样溶液进行顶空气相色谱与质谱联用测定;
12.其中,顶空气相色谱的顶空条件如下:顶空平衡温度为45℃~55℃;取样针温度为
50℃~60℃;顶空加热时间为8min~12min;进样体积为0.3ml~0.8ml;
13.顶空气相色谱的气相色谱条件如下:色谱柱温度为:35℃~45℃下保持1.5min~2.5min,以15℃/min~25℃/min的速率升温至55℃~65℃,保持1.5min~2.5min,再以45℃/min~55℃/min的速率升温至190℃~210℃,保持1.5min~2.5min;载气为氦气;进样口温度为190℃~210℃;分流比为(8~12):1;柱流速为1.2ml/min~1.8ml/min。
14.在其中一个实施例中,所述第一碱性溶液的组分包括第一碱与水,所述第二碱性溶液的组分包括第二碱与水;
15.所述第一碱与所述第二碱分别独立地选自氢氧化钠与氢氧化钾中的至少一种。
16.在其中一个实施例中,所述第一碱性溶液中,所述第一碱的浓度为0.8g/l~1.2g/l;和/或
17.所述第二碱性溶液中,所述第二碱的浓度为0.2g/l~0.6g/l。
18.在其中一个实施例中,所述第一酸性溶液的组分包括第一酸与水,所述第二酸性溶液的组分包括第二酸与水;
19.所述第一酸与所述第二酸分别独立地选自磷酸、盐酸与硫酸中的至少一种。
20.在其中一个实施例中,在所述第一酸性溶液与所述第二酸性溶液中,所述第一酸与所述第二酸的浓度分别独立地选自0.1ml/ml~0.3ml/ml。
21.在其中一个实施例中,所述第一氯胺t溶液的组分包括氯胺t与水,所述第二氯胺t溶液的组分包括氯胺t与水;
22.所述第一氯胺t溶液与所述第二氯胺t溶液中,氯胺t的浓度分别独立地选自5g/l~15g/l。
23.在其中一个实施例中,质谱条件如下:电离方式为电子轰击电离源;离子源温度为220℃~240℃;接口温度为210℃~230℃;溶剂延迟时间为0.8min~1.2min;监测方式为选择离子扫描(sim);扫描离子m/z为61、63与65;定量离子m/z为61。
24.在其中一个实施例中,所述含氰离子的标准品为水中氰成分分析标准物质。
25.在其中一个实施例中,将含氰离子的标准品与第一碱性溶液混合后还包括如下步骤:将混合后的物料与所述第二碱性溶液混合,制备中间标准品溶液。
26.在其中一个实施例中,所述中间标准品溶液中,所述氰离子的浓度为0.001mg/l~0.2mg/l。
27.与现有技术相比较,本发明的白酒或白酒发酵过程样本中氰化物含量的检测方法具有如下有益效果:
28.本发明采用顶空-气相色谱法与质谱联用测定白酒或白酒发酵过程样本中氰化物的含量,通过对待测物进行预处理,并限定顶空条件以及气相色谱条件,避免白酒以及白酒发酵过程样本中高浓度、高含量的乙醇以及低沸点的基质对检测产生影响,最终获得的检测结果就有较好的准确性、精密度以及较低的检测限。
附图说明
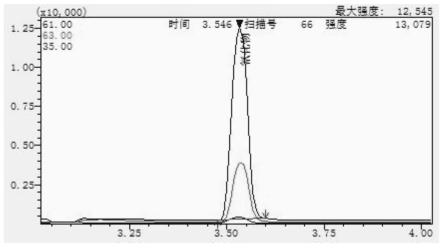
29.图1为本发明提供的标准品溶液的色谱图;
30.图2为本发明提供的标准品溶液的质谱图。
具体实施方式
31.为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施方式。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明的公开内容的理解更加透彻全面。
32.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
33.本发明提供了一种白酒或白酒发酵过程样本中氰化物含量的检测方法,包括如下步骤:
34.将含氰离子的标准品与第一碱性溶液混合,然后与第一酸性溶液混合,再与第一氯胺t溶液混合,制备标准品溶液;
35.将待测样品与第二碱性溶液混合,然后与第二酸性溶液混合,再与第二氯胺t溶液混合,制备试样溶液;
36.将标准品溶液与试样溶液进行顶空气相色谱与质谱联用测定;
37.其中,顶空气相色谱的顶空条件如下:顶空平衡温度为45℃~55℃;取样针温度为50℃~60℃;顶空加热时间为8min~12min;进样体积为0.3ml~0.8ml;
38.顶空气相色谱的气相色谱条件如下:色谱柱温度为:35℃~45℃下保持1.5min~2.5min,以15℃/min~25℃/min的速率升温至55℃~65℃,保持1.5min~2.5min,再以45℃/min~55℃/min的速率升温至190℃~210℃,保持1.5min~2.5min;载气为氦气;进样口温度为190℃~210℃;分流比为(8~12):1;柱流速为1.2ml/min~1.8ml/min。
39.在一个具体的示例中,第一碱性溶液的组分包括第一碱与水,第二碱性溶液的组分包括第二碱与水;
40.第一碱与所述第二碱分别独立地选自氢氧化钠与氢氧化钾中的至少一种。
41.更具体地,第一碱为氢氧化钠。
42.更具体地,第二碱为氢氧化钠。
43.在一个具体的示例中,第一碱性溶液中,第一碱的浓度为0.8g/l~1.2g/l。
44.可以理解地,在本发明中,第一碱性溶液中,第一碱的浓度包括但不限于0.8g/l、0.85g/l、0.9g/l、0.95g/l、1.0g/l、1.05g/l、1.1g/l、1.15g/l、1.2g/l。
45.在一个具体的示例中,第二碱性溶液中,第二碱的浓度为0.2g/l~0.6g/l。
46.可以理解地,在本发明中,第二碱性溶液中,第二碱的浓度包括但不限于0.2g/l、0.25g/l、0.3g/l、0.35g/l、0.4g/l、0.45g/l、0.5g/l、0.55g/l、0.6g/l。
47.在一个具体的示例中,第一酸性溶液的组分包括第一酸与水,第二酸性溶液的组分包括第二酸与水;
48.第一酸与第二酸分别独立地选自磷酸、盐酸与硫酸中的至少一种。
49.更具体地,第一酸为磷酸。更为具体地,磷酸为浓磷酸。
50.更具体地,第二酸为磷酸。更为具体地,磷酸为浓磷酸。
51.在一个具体的示例中,在第一酸性溶液与第二酸性溶液中,第一酸与第二酸的浓
度分别独立地选自0.1ml/ml~0.3ml/ml。
52.可以理解地,在本发明中,在第一酸性溶液与第二酸性溶液中,第一酸与第二酸的浓度包括但不限于0.1ml/ml、0.15ml/ml、0.2ml/ml、0.25ml/ml、0.3ml/ml。
53.在一个具体的示例中,第一氯胺t溶液的组分包括氯胺t与水,第二氯胺t溶液的组分包括氯胺t与水;
54.第一氯胺t溶液与第二氯胺t溶液中,氯胺t的浓度分别独立地选自5g/l~15g/l。
55.可以理解地,在本发明中,氯胺t是一种有机化合物,分子式为c7h7clnnao2s
·
3h2o,氯胺t的浓度包括但不限于5g/l、6g/l、7g/l、8g/l、9g/l、10g/l、11g/l、12g/l、13g/l、14g/l、15g/l。
56.当配制氯胺t溶液浑浊时,需更换新配制的氯胺t。
57.可以理解地,在本发明中,顶空平衡温度包括但不限于45℃、46℃、47℃、48℃、49℃、50℃、51℃、52℃、53℃、54℃、55℃。优选地,顶空平衡温度为50℃。
58.可以理解地,在本发明中,取样针温度包括但不限于50℃、51℃、52℃、53℃、54℃、55℃、56℃、57℃、58℃、59℃、60℃。优选地,取样针温度为55℃。
59.可以理解地,在本发明中,顶空加热时间包括但不限于8min、9min、10min、11min、12min。优选地,顶空加热时间为10min。
60.可以理解地,在本发明中,进样体积包括但不限于0.3ml、0.35ml、0.4ml、0.45ml、0.5ml、0.55ml、0.6ml、0.65ml、0.7ml、0.75ml、0.8ml。优选地,进样体积为0.5ml。
61.在一个具体的示例中,色谱柱温度为:40℃下保持2min,以20℃/min的速率升温至60℃,保持2min,再以50℃/min的速率升温至200℃,保持2min。
62.可以理解地,在本发明中,进样口温度包括但不限于190℃、191℃、192℃、193℃、194℃、195℃、196℃、197℃、198℃、199℃、200℃、201℃、202℃、203℃、204℃、205℃、206℃、207℃、208℃、209℃、210℃。优选地,进样口温度为200℃。
63.可以理解地,在本发明中,分流比包括但不限于8:1、9:1、10:1、11:1、12:1。优选地,分流比为10:1。
64.可以理解地,在本发明中,柱流速包括但不限于1.2ml/min、1.3ml/min、1.4ml/min、1.5ml/min、1.6ml/min、1.7ml/min、1.8ml/min。优选地,柱流速为1.5ml/min。
65.在一个具体的示例中,色谱柱选自毛细管柱。更具体地,毛细管柱为聚乙二醇固定相。更具体地,色谱柱的规格为:50米长,0.25mm内径,以及0.25μm膜厚。
66.在一个具体的示例中,质谱条件如下:电离方式为电子轰击电离源(ei);离子源温度为220℃~240℃;接口温度为210℃~230℃;溶剂延迟时间为0.8min~1.2min;监测方式为选择离子扫描;扫描离子m/z为61、63与65;定量离子m/z为61。
67.可以理解地,在本发明中,离子源温度包括但不限于220℃、221℃、222℃、223℃、224℃、225℃、226℃、227℃、228℃、229℃、230℃、231℃、232℃、233℃、234℃、235℃、236℃、237℃、238℃、239℃、240℃。优选地,离子源温度为230℃。
68.可以理解地,在本发明中,接口温度包括但不限于210℃、211℃、212℃、213℃、214℃、215℃、216℃、217℃、218℃、219℃、220℃、221℃、222℃、223℃、224℃、225℃、226℃、227℃、228℃、229℃、230℃。优选地,接口温度为220℃。
69.可以理解地,在本发明中,溶剂延迟时间包括但不限于0.8min、0.9min、1.0min、
1.1min、1.2min。优选地,溶剂延迟时间为1.0min。
70.在一个具体的示例中,含氰离子的标准品为水中氰成分分析标准物质。具体地,水中氰成分分析标准物质为经国家认证并授予标准物质证书的标准品。
71.在一个具体的示例中,将含氰离子的标准品与第一碱性溶液混合后还包括如下步骤:将混合后的物料与第二碱性溶液混合,制备中间标准品溶液。
72.在一个具体的示例中,中间标准品溶液中,氰离子的浓度为0.001mg/l~0.2mg/l。
73.以下结合具体实施例对本发明的白酒或白酒发酵过程样本中氰化物含量的检测方法做进一步详细的说明。以下实施例中所用的原料,如无特别说明,均为市售产品。除非另有说明,本方法所用试剂均为分析纯,水为gb/t 6682规定的二级水。
74.实施例1
75.本实施例提供一种白酒中氰化物含量的检测方法,具体如下:
76.一、试剂配制:
77.第一碱性溶液(0.1%氢氧化钠溶液):称取1.0g氢氧化钠,用水溶解定容至1l。
78.第二碱性溶液(0.01mol/l氢氧化钠溶液):称取0.4g氢氧化钠,用水溶解定容至1l。
79.第一酸性溶液与第二酸性溶液(磷酸溶液):量取10ml浓磷酸,加入到50ml水中,混合均匀。
80.第一氯胺t溶液与第二氯胺t溶液:称取0.1g氯胺t,用水溶解定容至10ml,现用现配。
81.二、制备标准品溶液:
82.(1)氰离子(以cn-计)标准中间溶液:准确移取2.00ml的水中氰成分分析标准物质(50.0μg/ml)于10.0ml的容量瓶,用0.1%氢氧化钠溶液定容,此溶液浓度为10.0mg/l,现用现配。
83.(2)氰离子(以cn-计)标准工作溶液:移取适量上述(1)制得的氰离子(以cn-计)标准中间溶液,用0.01mol/l氢氧化钠溶液稀释配制成浓度为0.001mg/l、0.005mg/l、0.050mg/l、0.100mg/l、0.200mg/l的工作溶液,现用现配。
84.(3)标准品溶液:分别准确移取10.0ml上述(2)制得的氰离子标准工作溶液于6个顶空瓶中,加入0.2ml磷酸溶液,然后加入0.2ml氯胺t溶液,立即加盖密封,涡旋混合,待测。
85.三、制备试样溶液:
86.(1)取白酒样品约500ml,充分混匀,装入洁净容器中,密封,于0℃~4℃条件下保存。
87.(2)准确移取0.20ml试样于顶空瓶中,加入0.01mol/l氢氧化钠溶液9.8ml,静置5min,加入0.2ml磷酸溶液,然后加入0.2ml氯胺t溶液,立即加盖密封,待测。
88.四、制备试样溶液:
89.除了不加白酒试样,其他操作均与上述第三点一致。
90.五、顶空气相色谱与质谱联用测定
91.(1)顶空分析条件如下:
92.顶空平衡温度:50℃;
93.取样针温度:55℃;
94.顶空加热时间:10min;
95.进样体积:0.5ml。
96.(2)气相色谱条件如下:
97.色谱柱:毛细管柱(聚乙二醇固定相),50m
×
0.25mm(内径)
×
0.25μm(膜厚);
98.色谱柱温度:40℃保持2min,以20℃/min速率升至60℃保持2min,再以50℃/min速率升至200℃保持2min;
99.载气:氦气,纯度≥99.999%;
100.进样口温度:200℃;
101.分流比:10:1;
102.柱流速:1.5ml/min。
103.(3)质谱条件如下:
104.电离方式:电子轰击电离源(ei);
105.离子源温度:230℃;
106.接口温度:220℃;
107.溶剂延迟时间:1min;
108.监测方式:选择离子扫描(sim),扫描离子m/z:61、63、35,定量离子m/z:61。
109.按照上述条件参数对标准品溶液进行顶空气相色谱与质谱联用测定,根据氰化物保留时间定性,测量样品溶液的峰面积(或峰高)响应值,采用外标法定量。样品溶液中氰化物衍生物的响应值应在标准线性范围内,若超出范围,在加磷酸溶液前用0.01mol/l氢氧化钠溶液稀释至范围内。
110.实施例2
111.本实施例提供一种白酒发酵过程样本中氰化物含量的检测方法,具体如下:
112.一、试剂配制:
113.第一碱性溶液(0.1%氢氧化钠溶液):称取1.0g氢氧化钠,用水溶解定容至1l。
114.第二碱性溶液(0.01mol/l氢氧化钠溶液):称取0.4g氢氧化钠,用水溶解定容至1l。
115.第一酸性溶液与第二酸性溶液(磷酸溶液):量取10ml浓磷酸,加入到50ml水中,混合均匀。
116.第一氯胺t溶液与第二氯胺t溶液:称取0.1g氯胺t,用水溶解定容至10ml,现用现配。
117.二、制备标准品溶液:
118.(1)氰离子(以cn-计)标准中间溶液:准确移取2.00ml的水中氰成分分析标准物质(50.0μg/ml)于10.0ml的容量瓶,用0.1%氢氧化钠溶液定容,此溶液浓度为10.0mg/l,现用现配。
119.(2)氰离子(以cn-计)标准工作溶液:移取适量上述(1)制得的氰离子(以cn-计)标准中间溶液,用0.01mol/l氢氧化钠溶液稀释配制成浓度为0.001mg/l、0.005mg/l、0.050mg/l、0.100mg/l、0.200mg/l的工作溶液,现用现配。
120.(3)标准品溶液:分别准确移取10.0ml上述(2)制得的氰离子标准工作溶液于6个顶空瓶中,加入0.2ml磷酸溶液,然后加入0.2ml氯胺t溶液,立即加盖密封,涡旋混合,待测。
121.三、制备试样溶液:
122.(1)取白酒发酵过程样本样品约500g,用样品粉碎装置将其制成粉末,装入洁净容器,密封,于0℃~4℃条件下保存。
123.(2)准确称取试样2g(精确至0.01g)于25ml比色管,用0.01mol/l氢氧化钠溶液定容至刻度,超声提取20min,转移至50ml离心管,4000r/min离心5min,然后准确移取2.5ml提取液于顶空瓶中,加入0.01mol/l氢氧化钠溶液7.5ml,静置5min,加入0.2ml磷酸溶液,涡旋混合,然后加入0.2ml氯胺t溶液,立即加盖密封,涡旋混合,待测。
124.四、制备试样溶液:
125.除了不加白酒试样,其他操作均与上述第三点一致。
126.五、顶空气相色谱与质谱联用测定
127.(1)顶空分析条件如下:
128.顶空平衡温度:50℃;
129.取样针温度:55℃;
130.顶空加热时间:10min;
131.进样体积:0.5ml。
132.(2)气相色谱条件如下:
133.色谱柱:毛细管柱(聚乙二醇固定相),50m
×
0.25mm(内径)
×
0.25μm(膜厚);
134.色谱柱温度:40℃保持2min,以20℃/min速率升至60℃保持2min,再以50℃/min速率升至200℃保持2min;
135.载气:氦气,纯度≥99.999%;
136.进样口温度:200℃;
137.分流比:10:1;
138.柱流速:1.5ml/min。
139.(3)质谱条件如下:
140.电离方式:电子轰击电离源(ei);
141.离子源温度:230℃;
142.接口温度:220℃;
143.溶剂延迟时间:1min;
144.监测方式:选择离子扫描(sim),扫描离子m/z:61、63、35,定量离子m/z:61。
145.按照上述条件参数对标准品溶液进行顶空气相色谱与质谱联用测定,根据氰化物保留时间定性,测量样品溶液的峰面积(或峰高)响应值,采用外标法定量。样品溶液中氰化物衍生物的响应值应在标准线性范围内,若超出范围,在加磷酸溶液前用0.01mol/l氢氧化钠溶液稀释至范围内。
146.对比例1:
147.本对比例提供一种白酒发酵过程样本中氰化物含量的检测方法,具体如下:
148.样品处理:准确称取经粉碎均匀的试样1g(精确至0.01g)于50ml离心管中,加入4ml蒸馏水,加盖密封,涡旋振荡器中震荡2min,再超声提取20min,然后10000rpm离心10min,准确移取2ml上清液于顶空瓶中,用水定容至10ml,加入0.2ml磷酸溶液,然后加入0.1ml氯胺t溶液,立即加盖密封,漩涡混合,待测。浓度超过线性范围应经适当稀释后重新
测定。
149.气相色谱柱条件如下:
150.hp-ffap石英毛细管柱(30m
×
0.32mm,0.25μm)。柱温:40℃保持5min,以50℃/min的速率升至240℃,保持2min。流速1.2ml/min,载气为高纯氮气,进样口温度250℃,分流进样,分流比10:1,电子捕获检测器温度300℃。
151.顶空条件如下:
152.样品平衡30min,样品平衡温度50℃,样品环温度为80℃,传输线温度95℃,进样持续0.03min,加压1min,gc循环20min。
153.质谱条件与实施例2一致。
154.对实施例1~2以及对比例1提供的白酒与白酒发酵过程样本中氰化物含量的检测方法进行评价:
155.1、浓度为0.05mg/l的标准品溶液的色谱图如图1所示,其质谱图如图2所示。标准品溶液和试样溶液的目标化合物在相同保留时间处出现,并且对应质谱碎片离子的质荷比与标准工作溶液的质谱图一致,其丰度比与浓度相当的标准工作溶液相比符合表1,可定性目标化合物。
156.表1气相色谱-质谱定性确证相对离子丰度最大容许误差
157.相对离子丰度(%)k》5050≥k》2020≥k》10k≤10允许的相对偏差(%)
±
10
±
15
±
20
±
50
158.2、白酒中氰化物(以cn-计)结果计算
159.试样中氰化物(以cn-计)含量按式(1)计算:
[0160][0161]
式中:
[0162]
x——试样中氰化物(以cn-计)的含量,单位为毫克每升(mg/l);
[0163]
ρ——由标准曲线得到的样液中氰化物的浓度,单位为毫克每升(mg/l);
[0164]
ρ0——由标准曲线得到的空白试验中氰化物的浓度,单位为毫克每升(mg/l);
[0165]
v——样品体积,单位为毫升(ml);
[0166]
10——加酸衍生前顶空瓶中溶液体积,单位为毫升(ml)。
[0167]
注:氰化物含量以hcn计时,结果乘以换算系数1.0385。
[0168]
计算结果保留三位有效数字。
[0169]
3、白酒发酵过程样本中氰化物(以cn-计)结果计算
[0170]
试样中氰化物(以cn-计)含量按式(2)计算:
[0171][0172]
式中:
[0173]
x——试样中氰化物(以cn-计)的含量,单位为毫克每千克(mg/kg);
[0174]
ρ——由标准曲线得到的样液中氰化物的浓度,单位为毫克每升(mg/l);
[0175]
ρ0——由标准曲线得到的空白试验中氰化物的浓度,单位为毫克每升(mg/l);
[0176]v1
——样品定容体积,单位为毫升(ml);
[0177]v2
——衍生用样液体积,单位为毫升(ml);
[0178]
m——样品质量,单位为克(g);
[0179]
10——加酸衍生前顶空瓶中溶液体积,单位为毫升(ml);
[0180]
1000——单位换算系数。
[0181]
说明:氰化物含量以hcn计时,以氰化物(以cn-计)结果乘以换算系数:1.0385。
[0182]
计算结果保留三位有效数字。
[0183]
评价结果如表2所示。
[0184]
表2
[0185][0186]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0187]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1