含维生素D3的制剂中有关物质的高效液相检测方法及供试品溶液的制备方法与流程
含维生素d3的制剂中有关物质的高效液相检测方法及供试品溶液的制备方法
技术领域
1.本发明涉及药物分析技术领域,具体涉及一种含维生素d3的制剂中有关物质的高效液相检测方法及高效液相检测方法中所用的供试品溶液的制备方法。
背景技术:
2.维生素d3是一种脂溶性维生素,也被看作是一种作用于钙、磷代谢的激素前体。常与碳酸钙、维生素a或多种维生素和矿物质组合在一起形成复方制剂,常见的含维生素d3的制剂有碳酸钙d3片、碳酸钙d3咀嚼片、碳酸钙d3颗粒剂、维d钙咀嚼片、儿童维d钙咀嚼片、碳酸钙d3元素片、维生素ad软胶囊等,其中维生素d3的含量为30iu-245iu。
3.维生素d3遇光或空气均易变质,中国药典规定的维生素d3原料药贮存方式为遮光、充氮、密封、在冷处保存。由于维生素d3遇光或空气易氧化和光解生成前维生素d3、反式维生素d3、光甾醇3和速甾醇3等杂质,在进行制剂研究时,如何保证维生素d3稳定是制剂厂家首要解决的问题,故越来越多制剂厂家采用维生素d3包合物作为原料进行制剂研究。维生素d3包合物由于包和的特殊性,在进行提取前首先需要将包合物溶解,使维生素d3分散出来,选择的预处理溶剂应能溶解包合物且不破坏维生素d3,同时还应具备溶解维生素d3的能力。现有技术中针对样品的处理流程较为繁琐,增加了有效成分的损耗,且提取率低、重现性差,进一步导致检测灵敏度降低。
4.目前药典方法仅公布了维生素d3原料有关物质检测方法,含色谱条件和供试品溶液配制方法,部分专利或文献公布了碳酸钙d3咀嚼片供试品制备方法及正相系统检测有关物质的方法,这些方法经申请人重现,发现由于供试品溶液制备过程中提取率低,导致检测的平行样本重现性差。
5.现有技术申请公布号为cn109709237a的发明专利,公开了一种碳酸钙d3咀嚼片的有关物质检测方法,具体公开了供试品溶液的制备方法和液相检测方法,该提取溶液的配制方法繁琐,使用了多种毒性的有机溶剂,实验人员操作风险高。同时,申请人发现有效成份不能提取完全、重现性差、灵敏度低。
6.现有技术中申请公布号为cn111239272a的发明专利,公开了一种检测含维生素d3的制剂中有关杂质的方法,具体公开了供试品溶液的制备方法和液相检测方法。其专利说明书中未公开制剂中维生素d3的含量,申请人尝试将该方法用于维生素d3含量较低的碳酸钙d3咀嚼片(维生素d3的含量为125iu)或碳酸钙d3片(维生素d3的含量为60iu)的有关物质检测,发现提取率低,每次提取的提取率差异大,重现性差、灵敏度低。另外,该提取方法中乙醇、水和正己烷三者直接混合,由于乙醇和正己烷可互溶,这三者混合很难分层,即使实现了分层,也难以萃取完全。并且提取和萃取同步进行,还可能造成提取和萃取均不完全,影响最终的测定结果。
7.由于人体单次对钙的吸收有限,因此营养学家建议钙补充剂适宜小剂量分多次服用,以促进人体更好的吸收,因此含低剂量的维生素d3的制剂具有很好的应用前景,但是现
有技术中没有专门针对含低剂量的维生素d3的制剂中有关物质的检测方法,无法很好的控制其质量,因此,开发一种针对含维生素d3的制剂的专属性强、重现性良好、灵敏度高的有关物质的检测方法及一种提取率高的供应品溶液制备方法尤为迫切。
技术实现要素:
8.本发明旨在克服上述现有技术的至少一种缺陷,提供一种用于检测含维生素d3的制剂中有关物质的供试品溶液的制备方法,以达到提取率高、重现性好、提取溶剂杂质少的目的。
9.本发明的另一目的在于,提供一种含碳酸钙和维生素d3的复方制剂有关物质的高效液相检测方法,以达到检测灵敏度高、专属性强的目的。
10.其中所述含维生素d3的制剂中的维生素d3含量≥31.25iu。优选的,所述制剂中维生素d3含量为31.25~200iu,更优选的,所述制剂中维生素d3含量为31.25iu、60iu、100iu、125iu或200iu。
11.具体的方案如下:
12.一种用于检测含维生素d3的制剂中有关物质的供试品溶液的制备方法,所述制备方法包括以下步骤:s1:提取:避光操作,称取维生素d3制剂适量,置棕色锥形瓶中,加入甲醇水溶液适量进行提取,并振摇,得提取液;s2:萃取:精密加入正己烷适量于所述提取液中,振摇后转移至分液漏斗,静置待分层,取上层正己烷层,转移至烧瓶中,向下层中继续添加适量正己烷,重复此萃取过程多次,将多次萃取得到的正己烷溶液合并在同一烧瓶中,得正己烷萃取液;s3:浓缩:45℃~60℃水浴条件下,对所述正己烷萃取液进行浓缩,并在旋转蒸发仪下蒸干;得供试品残渣;s4:配制:加0.5~2ml所述甲醇水溶液使所述供试品残渣溶解,经玻璃纤维滤膜过滤,得供试品溶液;其中,所述维生素d3制剂的规格≥31.25iu。
13.进一步的,所述步骤s1中,所述甲醇水溶液为90%~95%甲醇水溶液。
14.优选的,所述甲醇水溶液为95%甲醇水溶液。
15.本发明的一种实施方式为,所述甲醇水溶液为90%甲醇水溶液。
16.进一步的,所述甲醇水溶液的加入量为30~100ml。
17.优选的,所述甲醇水溶液的加入量为50~100ml。
18.进一步的,所述正己烷的加入量为10~100ml/次,萃取次数为2~10次。
19.优选的,所述正己烷的加入量为40~60ml/次,萃取次数为3~5次。
20.进一步的,所述步骤s1中,称取的样品中含有维生素d3的量≥20μg。优选的,所述步骤s1中,称取的样品中含有维生素d3的量为30μg。
21.进一步的,所述步骤s1中,所述提取的方式为超声提取或者水浴提取。
22.优选的,所述超声提取的功率为60~100w,超声时间为5~30min;所述水浴提取条件为45
±
5℃水浴提取。
23.进一步的,所述步骤s1中,所述振摇的频次为1~2min摇一次,振摇方式为手动或者机械振摇。
24.所述振摇可以是手动振摇或者机械振摇,申请人经过对比发现,在提取过程中,每隔1~2min振摇一次,能够明显提高提取率。
25.进一步的,所述步骤s3中,所述水浴的温度为50~60℃。
26.进一步的,所述含维生素d3的制剂为包含碳酸钙和维生素d3的制剂。
27.优选的,所述制剂为碳酸钙d3咀嚼片、碳酸钙d3咀嚼片(ii)、维d钙咀嚼片、儿童维d钙咀嚼片、碳酸钙d3颗粒、复方碳酸钙颗粒、或复方碳酸钙泡腾颗粒、碳酸钙d3片、碳酸钙维d3元素片或维生素d3片。
28.更为优选的,所述制剂为碳酸钙d3咀嚼片(ii),其中维生素d3的含量为60iu。
29.本发明又一优选方案针对的制剂为碳酸钙d3片,其中维生素d3的含量为125iu。
30.所述咀嚼片为碳酸钙d3咀嚼片、碳酸钙d3咀嚼片(ii)或维d钙咀嚼片,其中维生素d3的含量为60iu~200iu,具体的,碳酸钙d3咀嚼片中维生素d3的含量为200iu;碳酸钙d3咀嚼片(ii)中维生素d3的含量为60iu;维d钙咀嚼片中维生素d3的含量为100iu。
31.本发明的另一优选方案为,所述颗粒剂为碳酸钙d3颗粒、复方碳酸钙颗粒或复方碳酸钙泡腾颗粒,其中维生素d3的含量为31.25iu~125iu。具体的,碳酸钙d3颗粒中维生素d3的含量为100iu;复方碳酸钙颗粒中维生素d3的含量62.5iu;复方碳酸钙泡腾颗粒中维生素d3的含量为31.25iu或125iu。
32.本发明的另一优选方案为,所述片剂为碳酸钙d3片、碳酸钙维d3元素片或维生素d3片,其中维生素d3的含量为100iu~200iu。具体的,碳酸钙d3片或碳酸钙维d3元素片中维生素d3的含量为125iu;维生素d3片中维生素d3的含量为200iu。
33.本发明提供一种含维生素d3的制剂中有关物质的高效液相检测方法,所述方法包括以下步骤:
34.a)制备供试品溶液:按照上述制备方法制备得到供试品溶液;
35.b)制备对照品溶液:精密称取维生素d3对照品适量,用甲醇溶解并定量稀释制成每1ml中含维生素d
3 0.2μg的溶液,作为对照品溶液;
36.c)液相色谱检测:精密量取供试品溶液和对照品溶液,分别注入液相色谱仪,记录色谱图。
37.发明人经过大量的筛选实验发现,利用本发明的提取方法提取率可接近100%,而参考现有技术中的提取方法,由于提取条件不佳,平行实验之间的差异可能超过一倍,最终提取率低,重现性非常差。发明人针对维生素d3的提取方法进行了全面的考察,最终筛选出的提取方法重现性好、提取率高。在液相色谱检测方面,参考现有技术中的正相色谱检测、或者c18色谱柱的反相色谱法均可实现含维生素d3的制剂中有关物质的检测。
38.优选的,所述色谱条件为:色谱柱固定相填充剂为十八烷基硅烷键合硅胶;流动相a:0.01%~0.5%磷酸溶液;流动相b:乙腈-流动相a(90~99∶1~10)梯度洗脱;检测波长为265nm;进样体积为50~250μl。
39.发明人对比了正相高效液相色谱检测和反相高效液相色谱检测,发现利用反相高效液相色谱法进行检测,检测的峰形更加标准,主峰和相邻峰之间、已知杂质与相邻峰之间的分离度更好,且检测的杂质种类更多、更加全面,因此,优选采用反相液相色谱方法进行检测。
40.进一步的,所述步骤c)中,流动相的配制为:流动相a:0.025%~0.05%磷酸溶液;流动相b:乙腈-流动相a(99∶1);检测波长为265nm;进样体积为100~200μl;柱温为25~30℃;流速为0.5~1.5ml/min。
41.更为优选的,所述色谱条件:柱温为25℃;流速为0.7~1.0ml/min。
42.进一步的,所述步骤c)中,所述色谱柱为thermoaccucore xl、phenomenex utremex
tm c18 80a 或phenosphere 80a ods,内径
×
柱长为4.6
×
(150~250)mm,色谱柱的粒径3~5μm;洗脱条件选自以下洗脱程序中的一种:时间(min)流动相a%流动相b%015858015858501001450100145.115851551585
43.或:时间(min)流动相a%流动相b%01090501090609010150901015110901601090
44.或:时间(min)流动相a%流动相b%051.548.516138739109043010057010057.151.548.56551.548.5
45.优选的,所述色谱柱为thermoaccucore xl,内径
×
柱长为4.6
×
250mm,色谱柱的粒径4μm,洗脱程序为:时间(min)流动相a%流动相b%015858015858501001450100145.115851551585
46.另一优选方案为,所述色谱柱为phenomenex utremex
tm c18 80a,内径
×
柱长为4.6
×
250mm,色谱柱的粒径5μm,梯度洗脱程序如下:时间(min)流动相a%流动相b%
01090501090609010150901015110901601090
47.另一优选方案为,所述色谱柱为phenosphere 80a ods,内径
×
柱长为4.6
×
150mm,色谱柱的粒径3μm;梯度洗脱程序如下:时间(min)流动相a%流动相b%051.548.516138739109043010057010057.151.548.56551.548.5
48.优选的,所述含维生素d3的制剂包含碳酸钙和维生素d3。
49.进一步的,所述制剂为碳酸钙d3咀嚼片、碳酸钙d3咀嚼片(ii)、维d钙咀嚼片、儿童维d钙咀嚼片、碳酸钙d3片、复方碳酸钙颗粒或复方碳酸钙泡腾颗粒等维生素d3的含量为31.25iu~200iu的制剂。
50.优选的,所述复方制剂中维生素d3的含量为31.25~200iu。更优选的,维生素d3的含量为31.25iu、60iu、62.5iu、100iu、125iu或200iu。
51.需要说明的是,利用本发明的提取方法和检测方法能够实现低剂量的维生素d3的制剂中有关物质的检测,提取率高、灵敏度高、重现性好和具有高专属性;专业技术人员知晓,本发明的提取方法和有关物质检测方法同样适用于更高含量的维生素d3的制剂,因此针对含维生素d3的制剂中有关物质检测,只要运用了本发明公开的提取方法和/或检测方法,则均在本发明的保护范围之内。
52.本发明的有益效果:
53.(1)本发明提供了一种提取率高的供试品溶液的制备方法,该方法进行了充分的提取率实验考察,结果显示,各提取步骤均已提取完全,重现性好,适于推广应用。
54.(2)本发明供试品溶液的制备过程中用到的有机溶剂种类较少,毒性相对较低,进一步减少了实验人员的操作风险。
55.(3)本发明首次采用反相高效液相色谱法对含低剂量的维生素d3的制剂中有关物质进行检测,相比现有技术中的正相检测系统,其专属性更强、重现性更好、灵敏度更高,采用本发明的梯度洗脱的方式,各主峰与杂质峰均可被检出并良好分离,并且主峰的响应度高。
56.(4)本发明有关物质的检测方法能够有效分离和测定出低至31.25iu的复方碳酸钙泡腾颗粒或低至60iu的碳酸钙d3咀嚼片(ii)中维生素d3的杂质,可有效控制含维生素d3的复方制剂的质量,特别是低剂量碳酸钙d3咀嚼片的质量。
附图说明
57.图1为实施例2中系统适用性溶液hplc图谱。
58.图2为实施例2中维生素d3原料药供试品溶液hplc图谱。
59.图3为实施例2中碳酸钙d3片供试品溶液hplc图谱。
60.图4为实施例2中维生素d3杂质定位图谱。
61.图5为实施例2中的供试品溶液与空白对照hplc对比图谱。
62.图6为实施例2中的不同碳酸钙d3片产品hplc对比图谱。
63.图7为实施例3中的供试品溶液和系统适用性溶液hplc对比图谱。
64.图8为实施例4中检测方法重现性考察的hplc图谱。
65.图9为对比例1中检测方法重现性考察的hplc图谱。
具体实施方式
66.为使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施方式,对本发明进行进一步的详细说明。应当理解的是,此处所描述的具体实施方式仅用以解释本发明,并不限定本发明的保护范围。实施例1
1.本实施例用于考察供试品溶液制备方法的供试品提取率以及重现性。
2.1)色谱条件
3.仪器:高效液相色谱仪;
4.色谱柱:c18柱,phenomenex utremex
tm c18 80a,4.6mm
×
250mm,5μm;
5.流动相:乙腈-甲醇(91:9);
6.检测波长:265nm;
7.柱温:27℃;
8.流速:0.7ml/min;
9.2)样品制备
10.供试品溶液制备:
11.供试品:避光操作,取相当于含维生素d
3 30μg的碳酸钙d3咀嚼片(ii)研细粉末(其中碳酸钙d3咀嚼片(ii)中维生素d3的含量为60iu),置棕色锥形瓶中,加入95%甲醇水溶液适量,超声提取30min,1min手动振摇一次,精密加入正己烷30ml,振摇后,转移至分液漏斗,静置待分层,取上层清液即正己烷层,转移至烧瓶中,向下层中继续添加正己烷30ml,重复此萃取过程4次,将全部正己烷溶液合并至同一烧瓶中,在50℃~60℃水浴并在旋转蒸发仪中蒸干,残渣加95%甲醇水溶液1ml使溶解,经玻璃纤维滤膜过滤,取续滤液作为供试品溶液。
12.对照品溶液制备:精密称取维生素d3对照品,用甲醇溶解并定量稀释制成每1ml中约含维生素d
3 0.2μg的溶液,作为对照品溶液;
13.3)实验结果
14.本实验设有两组平行对照,其中样品1和样品2为供试品经过甲醇水溶液超声提取后得到的甲醇水提取液样品;样品3和样品4分别为样品1和样品2经过正己烷萃取后甲醇水溶液层样品;样品5和样品6为最终得到的供试品溶液。
15.由下表可知,样品1、样品2、样品3和样品4的提取率均在98%以上,说明甲醇水溶液能将碳酸钙d3咀嚼片中的维生素d3基本溶解完全;且经过正己烷的多次萃取之后,甲醇水层中基本检测不到维生素d3,而最终得到的供试品溶液中提取率均在99%以上,说明本发明选用的甲醇水溶液对供试品中维生素d3溶解完全,且采用正己烷进行萃取,萃取充分;因此本发明的提取方法对维生素d3的提取率高,且两组平行试验得到的趋势相同,说明本发明的提取方法重现性好。表1为本发明检测方法提取率及重现性考察结果
样品编号称样量(g)理论浓度μg/ml峰面积实测浓度μg/ml残留率/提取率(%)124.0148610.30561063280.300598.3224.0185580.31821133650.3204100.7324.0148610.3056ndn/an/a424.0185580.3182ndn/an/a524.01486119.5581686170719.393899.2624.01855820.3650731948520.6876101.6
实施例2
16.本实施例为针对本发明的检测方法的专属性和灵敏度的考察。
17.1)色谱条件
18.仪器:高效液相色谱仪;
19.色谱柱:c18柱,thermoaccucore xl,4.6
×
250mm,4μm
20.流动相:流动相a:0.05%磷酸溶液;流动相b:乙腈-流动相a(99:1);检测波长:265nm;
21.进样体积:100μl;
22.样品盘控温为5℃
±
3℃
23.柱温:25℃;
24.梯度条件:时间(min)流动相a%流动相b%015858015858501001450100145.11585155/1951585
25.2)样品制备
26.供试品溶液制备:避光操作,取相当于含维生素d
3 45μg的碳酸钙d3片(其中维生素d3的含量为125iu)研碎的颗粒,置棕色锥形瓶中,加入90%甲醇水溶液适量,超声提取10min,并不时振摇,精密加入正己烷100ml,振摇后,转移至分液漏斗,静置待分层,取上层清液即正己烷层,转移至烧瓶中,向下层中继续添加正己烷60ml,重复此萃取过程2次,将全部正己烷溶液合并至同一烧瓶中,在50℃~60℃水浴并在旋转蒸发仪中蒸干,残渣加90%甲醇水溶液2ml使溶解,经玻璃纤维滤膜过滤,取续滤液作为供试品溶液;
27.空白辅料溶液制备:按照上述供试品溶液制备方法制备得到;
28.对照品溶液制备:精密称取维生素d3对照品,用甲醇溶解并定量稀释制成每1ml中约含维生素d
3 0.2μg的溶液,作为对照品溶液;
29.灵敏度溶液制备:精密量取对照品溶液2ml,用溶剂定量稀释制成每1ml中约含维生素d
3 0.02μg的溶液,作为灵敏度溶液;
30.系统适用性溶液制备:用二氯甲烷配制每1ml中含维生素d
3 4.5mg的溶液,吸取10ml,在搅拌下缓慢地加入1,3-二环己基碳二亚胺20mg,继续搅拌1小时,立即精密量取1ml,置200ml量瓶中,加溶剂[甲醇:水(95∶5),下同]稀释至刻度,摇匀,将该溶液在70℃~75℃加热35分钟,冷却至室温,再将该溶液置uv光感稳定箱(26w/m2)或8.67w
·
hr/m2等效uv照射箱中照射5分钟。
31.3)实验结果
32.结果如图1~3所示,各图中峰a代表杂质a:反式维生素d3,峰b代表杂质b:前维生素d3;主峰c代表维生素d3,峰e代表杂质e:速甾醇3。其中图1为系统适用性溶液hplc图谱,图2是维生素d3原料药供试品溶液hplc图谱,图3为碳酸钙d3咀嚼片供试品溶液hplc图谱。
33.图4是维生素d3杂质定位图谱和系统适用性溶液图谱的对比图,由图谱可知,在该色谱条件下可检出所有已知杂质并能完全分离,与现有技术相比,本发明的提取方法结合检测方法,能够检测至少5种已知杂质,结合图4可知,包含杂质a(反式维生素d3)、杂质b(前维生素d3)、杂质c(光甾醇3)、杂质d(异速甾醇)、杂质e(速甾醇3)及主成分(维生素d3),各峰分离度好。
34.由图1可以看出,系统适用性溶液检测到了维生素d3:保留时间72.9min,反式维生素d3:保留时间46.4min,前维生素d3:保留时间58.6min,速甾醇:保留时间67.6min,如图2~3所示,选用本发明的供试品制备方法和液相检测方法,供试品溶液中杂质峰与主峰分离度高,杂质峰之间分离度高,原料药中检测到的各杂质峰与供试品溶液中检测到的杂质峰能够相互对应,说明辅料中的杂质与原料药中的杂质不会相互干扰,该分析方法适用性高。
35.图5为四种溶液的色谱图,其中空白溶液为甲醇水溶液;其中sys为系统适用性溶液;其中空白辅料溶液为利用碳酸钙d3片的辅料按照供试品溶液的制备方法制备得到;如图4~5所示,供试品溶液中检测到的维生素d3峰与相邻峰能完全分离;从系统适用性溶液和杂质定位溶液对比结果可知,已知杂质反式维生素d3、异速甾醇3、前维生素d3、速甾醇3、维生素d3、光甾醇3等杂质均能够被检测,且完全分离;结合空白溶液、空白辅料溶液检测结果可知,均没有干扰峰;说明本发明的检测方法专属性强。灵敏度溶液检测结果显示信噪比》10,按有关物质检查时浓度20μg/ml计算,其定量浓度百分比为0.1%。结果表明,该方法灵敏度高,可以充分满足有关物质检查测定的要求。
36.图6中对比了市面上现有的碳酸钙d3片(125iu)和自研的碳酸钙d3片(125iu)有关物质的检测,发现本发明的提取方法针对已上市的碳酸钙片和自研的碳酸钙片均能够提取完全,且本发明的检测方法均能实现上述制剂中有关物质的定量检测,各峰之间分离度良好。因此,本发明有关物质的检测方法能够有效分离和测定出碳酸钙d3片中维生素d3的杂质,可有效控制低剂量碳酸钙d3片的质量。实施例3
37.本实施例进一步证实本发明的检测方法测得的供试品溶液主峰和杂质峰分离度
高。
38.1)色谱条件
39.仪器:高效液相色谱仪;
40.色谱柱:c18柱,phenosphere 80a ods,4.6
×
150mm,3μm
41.流动相:流动相a:0.025%磷酸溶液;流动相b:乙腈-流动相a(99:1);检测波长:265nm;
42.进样体积:100μl;
43.样品盘控温为5℃
±
3℃;
44.柱温:25℃;
45.梯度条件:时间(min)流动相a%流动相b%051.548.516138739109043010057010057.151.548.56551.548.5
46.2)样品制备
47.供试品溶液制备:避光操作,取相当于含维生素d
3 45μg的碳酸钙维d3元素片研细颗粒,其中碳酸钙维d3元素片中维生素d3的含量为125iu,置棕色锥形瓶中,加入95%甲醇水溶液适量,超声提取20min,2min振摇一次,精密加入正己烷60ml,振摇后,转移至分液漏斗,静置待分层,取上层清液即正己烷层,转移至烧瓶中,向下层中继续添加正己烷20ml,重复此萃取过程8次,将全部正己烷溶液合并至同一烧瓶中,在50℃~60℃水浴并在旋转蒸发仪中蒸干,残渣加95%甲醇水溶液2ml使溶解,经玻璃纤维滤膜过滤,取续滤液作为供试品溶液;
48.对照品溶液制备:精密称取维生素d3对照品,用甲醇溶解并定量稀释制成每1ml中约含维生素d
3 0.2μg的溶液,作为对照品溶液;
49.系统适用性溶液:用二氯甲烷配制每1ml中含维生素d
3 4.5mg的溶液,吸取10ml,在搅拌下缓慢地加入1,3-二环己基碳二亚胺20mg,继续搅拌1小时,立即精密量取1ml,置200ml量瓶中,加溶剂[甲醇:水(95:5),下同]稀释至刻度,摇匀,将该溶液在70℃~75℃加热35分钟,冷却至室温,再将该溶液置uv光感稳定箱(26w/m2)或8.67w
·
hr/m2等效uv照射箱中照射5分钟。
50.3)试验结果
51.如图7所示,利用本实施例的检测方法,1为供试品溶液的检测图谱,2为系统适用性溶液的检测图谱;从系统适用性溶液的检测结果可知,杂质反式维生素d3、前维生素d3、速甾醇3等杂质均能够被检测,且分离度高;供试品溶液中主峰和相邻杂质峰分离度高,供试品溶液中没有其他杂峰干扰,说明本发明检测方法优。实施例4
1.本实施例是针对本发明所述的检测方法重现性的考察。
2.本实施例选用复方碳酸钙泡腾颗粒(其中维生素d3的规格为31.25iu),按照实施例3中系统适用性溶液和供试品溶液制备方法制备相关溶液,其中供试品溶液制备2份平行的供试品溶液,最终得到系统适用性溶液、供试品溶液1和供试品溶液2。
3.色谱检测条件如下:
4.仪器:高效液相色谱仪;
5.色谱柱:thermoaccucore xl,4.6
×
250mm,4μm
6.流动相:流动相a:0.01%磷酸溶液;流动相b:乙腈-流动相a(95:5);检测波长:265nm;
7.进样体积:100μl;
8.样品盘控温为5℃
±
3℃
9.柱温:25℃
10.梯度条件:时间(min)流动相a%流动相b%01090501090609010150901015110901551090
11.检测结果如图8所示,图8将系统适用性溶液、供试品溶液1、供试品溶液2的液相色谱图合为一张图谱,各主峰之间保留时间一致,为了更好地分析三个主峰的峰面积和峰高,发明人将三个主峰的位置进行了平行偏移,以便对比。其中1号峰为系统适用性溶液中的维生素d3主峰,2号峰为供试品溶液1中的维生素d3主峰,3号峰为供试品溶液2中的维生素d3主峰。如图8所示,平行制备的两份供试品溶液主峰峰高非常接近,说明本发明的提取率较为稳定,提取方法和检测方法重现性好。实施例5
12.本实施例与实施例1的不同之处在于,本实施例考察了提取过程中由超声提取改为水浴振荡对样品提取率的影响,具体供试品的制备方法为:避光操作,取相当于含维生素d330μg的碳酸钙d3咀嚼片(其中维生素d3的含量为200iu)研细的粉末,置棕色锥形瓶中,加入甲醇水(95:5)适量,45℃水浴提取30min,经玻璃纤维滤膜过滤,取续滤液进样检测。对比实施例1(超声振荡)的方法,对比结果如下表所示:表2水浴振荡的提取方式对提取率的影响
提取方法称样量(g)理论浓度μg/ml峰面积实测浓度μg/ml残留率/提取率(%)超声提取24.0148610.30561063280.300598.3水浴提取24.117450.62002094950.580095.4
13.由上述结果可知,水浴振荡的提取方式,其提取率也能超过95%,但是提取率稍低于超声提取,因此,优选的提取方式为超声提取。对比例1
14.本对比例采用现有技术中的样品提取方法和检测方法进行,具体方法如下:
15.1)色谱条件:
16.仪器:高效液相色谱仪;
17.色谱柱:supelcosiltm lc-si,4.6
×
150mm,3μm;
18.流动相:正己烷-正戊醇(997:3);
19.进样体积:100μl;
20.样品盘控温为5℃
±
3℃;
21.柱温:25℃;
22.洗脱条件:正己烷-正戊醇(997:3)等度洗脱;
23.2)供试品溶液制备:精密称取复方碳酸钙泡腾颗粒研细粉末(其中维生素d3的含量为31.25iu)适量(约相当于含维生素d
3 30μg的量),置50ml具塞离心管中,加二甲亚砜-水(3:1)混合液30ml和甲醇10ml,密塞,旋转振摇,50℃水浴振摇5分钟,精密加正己烷20ml,手腕式振摇20分钟,静置10分钟,将上层液体转移至50ml离心管中,以3000rpm转速离心10分钟,精密量取上清液10ml,置西林瓶中,用氮气流吹干,精密加正己烷1ml,使残渣完全溶解;经玻璃纤维滤膜过滤,取续滤液作为供试品溶液;按照该供试品溶液制备方法,制备两份平行的供试品溶液,命名为供试品溶液1和供试品溶液2。
24.系统适用性溶液:取维生素d3对照品约25mg,精密称定,置100ml棕色量瓶中,加异辛烷80ml,避免加热,超声处理1分钟使完全溶解,用异辛烷稀释至刻度,摇匀,充氮密塞,避光,0℃以下保存,作为对照品贮备溶液(1);精密量取5ml,置50ml棕色量瓶中,用异辛烷稀释至刻度,摇匀,充氮密塞,避光,0℃以下保存,作为对照品贮备溶液(2)。精密量取对照品贮备溶液(2)5ml,置具塞玻璃容器中,通氮后密塞,置90℃水浴中加热1小时,取出,迅速冷却,加正己烷5ml,摇匀,置lcm具塞石英吸收池中,在2支8w主波长分别为254nm和365nm的紫外光灯下,将石英吸收池斜放成45
°
并距灯管5/6cm,照射5分钟。
25.3)试验结果
26.由图9所示,1号峰为供试品溶液1主峰,2号峰为供试品溶液2主峰,3号峰为系统适用性溶液主峰,为了更好地分析三个主峰的峰面积和峰高,发明人将三个主峰的位置进行了平行偏移,以便对比。由图可知,平行制备的2份供试品溶液主峰面积差异大,说明本对比例中的样品提取方法无法提取完全,导致每次的提取率差异大,说明本方法重现性差。对比例2~4
1.对比例2~4针对的供试品为碳酸钙d3咀嚼片(ii)研磨后的细粉,其中碳酸钙d3咀嚼片(ii)中维生素d3的含量为60iu。
2.对比例3~4与实施例1的不同之处在于,对比例3~4采用了不同的样品处理方法,得到不同的供试品溶液,并考察了每份供试品溶液的提取率。
3.对比例2与实施例1的提取方法相同,即为实施例1步骤s1中的提取溶液,具体为:避光操作,取相当于含维生素d
3 30μg的供试品置棕色锥形瓶中,加入甲醇水(95:5)适量,超声提取30min,并不时振摇,经玻璃纤维滤膜过滤,取续滤液进样检测,得供试品溶液1。
4.对比例3与对比例2的不同之处在于,对比例3采用的是对比例1类似的方法制备供试品溶液,具体为:避光操作,精密称取供试品适量,约相当于含维生素d
3 30μg的量,置50ml具塞离心管中,加二甲亚枫-水(3:1)的混合液15ml与甲醇5ml,密塞,旋转振摇,置50℃
水浴保温5分钟,并不断振摇,精密加正己烷10ml,机器振摇20分钟,取正己烷层,重复萃取操作2次,每次加10ml正己烷,合并正己烷层,以3000rpm转速离心10分钟,精密量取上清液24ml,用氮气流吹干,精密加甲醇1ml,使残渣完全溶解,经玻璃纤维滤膜过滤,得供试品溶液2;
1.对比例4与对比例2的不同之处在于,对比例4中的提取溶剂为乙腈水(95:5),具体方法为:避光操作,取相当于含维生素d
3 30μg的供试品置棕色锥形瓶中,加入乙腈水(95:5)适量,超声提取30min,并不时振摇,经玻璃纤维滤膜过滤,取续滤液进样检测,得供试品溶液3。
2.试验结果如下表所示:表3为对比例2~对比例4的提取率考察结果
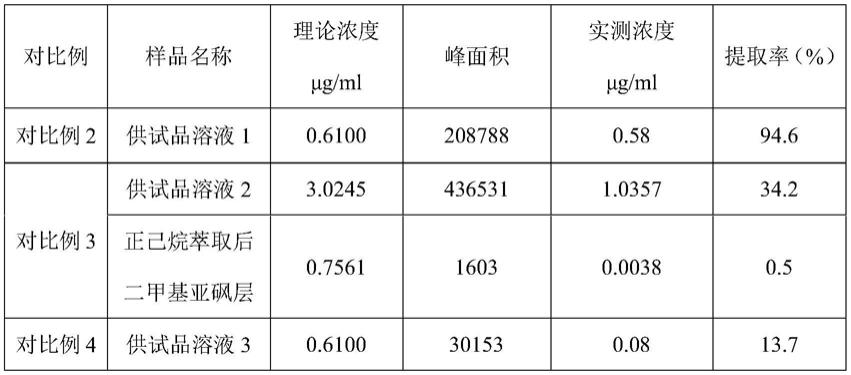
3.通过对实施例1的表1和对比例2~4的表3对比可知,对比例3采用与对比例1类似的方法进行提取,其中供试品溶液2的提取率仅34.2%,且正己烷萃取后二甲基亚砜层仍然检测到一定的维生素d3,说明正己烷的萃取也不完全,因此,结合对比例3的萃取前后溶液的检测,可知,该方法提取、萃取各个步骤均不完全,无法在实际检测中运用。
4.对比例4采用的是乙腈水溶液(95:5)提取,由上表可知,供试品溶液3提取率仅13.7%,提取溶剂的选择决定了供试品溶液的提取率和最终检测的重现性,因此,利用本发明的提取方法,优选甲醇水溶液为提取溶剂;更为优选的为95%甲醇水溶液为提取溶剂,能够实现较高的提取率。
5.根据以上对比例可知,由于现有技术中样品的提取方法不佳,提取率低,平行提取重现性差,灵敏度不高不适用于维生素d3含量更低(如31.25iu或60iu)的碳酸钙d3复方制剂的检测。
6.本发明申请人开发的供试品溶液制备方法和高效液相检测方法,所述供试品溶液制备方法使用的溶剂相对友好,提取率接近100%,经验证重现性好,可用作含量低至31.25iu的含维生素d3的固体制剂的含量测定和有关物质检测;所述高效液相色谱方法,专属性强、灵敏度高、重现性好,适用于含量低至31.25iu的含维生素d3的固体制剂的有关物质检测。
7.显然,本发明的上述实施例仅仅是为清楚地说明本发明技术方案所作的举例,而并非是对本发明的具体实施方式的限定。凡在本发明权利要求书的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1