重组人促红细胞生成素的含量检测方法与流程
1.本发明涉及生物检测技术领域,特别是涉及一种重组人促红细胞生成素的含量检测方法。
背景技术:
2.促红细胞生成素(简称促红素erythropoietin,epo),最早发现于1906年,是一种主要由肾脏近曲小管附近的细胞合成和分泌的糖蛋白,属唾液糖蛋白激素,蛋白质部分由166个氨基酸组成,分子量为34kd。促红细胞生成素依据其糖型结构的差异可分为α、β两种,α型含34%的碳水化合物,β型含26%的碳水化合物,两种类型在生物学特性、抗原性等效果上均相同。
3.1985年,人们利用基因重组技术表达了重组人促红细胞生成素(rhepo),应用dna重组技术将epo的基因构建到中国仓鼠卵巢细胞(cho细胞)中,形成可以表达rhepo基因的cho细胞,rhepo与人体内源性epo具有相同的生理活性。1989年6月,美国fda正式批准由安进公司研制的重组人促红细胞生成素上市。rhepo在临床上主要用于治疗慢性肾衰竭合并贫血症、慢性肾功能衰竭(crf)贫血、骨质增生异常综合征(mds)贫血、aids病人的伴生贫血等贫血的治疗,目前也有报道应用于治疗癌症病人接受化疗引起的贫血,纠正多发性骨髓瘤(mm)相关的贫血,早产儿贫血和孕妇妊娠期及产后贫血,类风湿关节炎、红斑狼疮及严重的寄生虫病患者的慢性贫血,血红蛋白病的镰状红细胞贫血及地中海贫血,炎症性肠疾患相关性贫血。同时,rhepo还可用于增强运动耐力,减少高原反应和需输血的心绞痛患者。总之,rhepo是全球最早的重组蛋白药物之一,也是技术成熟、疗效确切的基因工程药物。然而,在传统技术中,cho细胞表达rhepo的效率并不高,造成了rhepo整体存在产率低、成本高的问题,影响了其在临床上的推广。
4.为了提高rhepo的产率,需要优化cho细胞培养条件及下游纯化工艺,通过对细胞上清及中间品的rhepo含量进行定量检测,可以快速、准确地评估各项制备工艺参数的优劣势,实现提高rhepo产率的目的。
5.目前,用于rhepo含量检测的方法主要分为两类:一是生物学方法检测,包括生物体内测定法和生物体外测定法,由于生物学方法检测的是细胞因子的生物活性,因此具有高亲和力,但存在着特异性差、操作繁琐、易受干扰等缺点;二是免疫学方法检测,其是伴随免疫标记技术发展起来的检测方法,是目前应用最广泛检测rhepo的方法。目前的免疫学方法检测又分可为:放射免疫检测(ria)、酶联免疫吸附方法(elisa)、荧光免疫分析法(fia)、化学发光免疫测定(clta)等。
6.最初关于rhepo含量的检测基本上都采用ria方法。该方法技术成熟,灵敏度高,缺点是手工操作,出结果慢,试剂半衰期短,对环境有污染等,限制了该方法的应用。elisa是一种敏感的技术,可以在微克乃至纳克水平对所测物质进行定量,具有简便、成本低廉易于标准化等优点,缺点是样品用量大,每次检测需约200μl样品,检测时间长,人为误差较大。fia是非放射免疫法中灵敏度最高的,它常用来检测含量很低的生物活性物质,特异性和灵
敏度都较高,缺点是试剂盒昂贵,不适用于工艺摸索阶段使用。clta是以化学发光信号示踪藉以检测抗原或抗体的分析技术,其灵敏度一般均高于elisa,并且具有特异性强、标记物稳定、线性范围宽、检测速度快等优点。目前clia发展迅速,有取代ria而成为主流的趋势,但同时也存在检测系统价格昂贵难以普及,或价格便宜但是灵敏度和检测的可靠性得不到保证的不足。
7.有方法涉及一种基于磁珠为载体的促红细胞生成素检测法,通过在磁珠上偶联捕获抗体(抗epo抗体)捕获血清中的epo,随后加入生物素化的检测抗体,形成以磁珠为载体的“捕获抗体、epo、检测抗体”免疫复合物,然后加入链亲和素偶联的酶与检测抗体上标记的生物素结合,标记免疫复合物。该免疫分析方法,具有超高的灵敏度(19pg/ml),较传统的免疫分析方法提高13倍,样品用量(15μl)较传统免疫分析方法少13倍,样品检测实验周期缩短至34min,远短于传统免疫分析法(6h~8h)。然而,该方法虽然灵敏度高、样品用量少、检测周期短,但以磁珠为载体的“试剂”制作复杂,存在如需要制备抗rhepo抗体、难以形成商品化产品、“磁珠试剂”批次之间可能存在质量差异等问题,且检测过程过于繁琐,存在抗rhepo抗体脱落的风险,进而影响rhepo含量检测的准确性。
技术实现要素:
8.基于此,本发明提供一种检测准确度高、操作简单的重组人促红细胞生成素的含量检测方法。
9.具体技术方案如下:
10.一种重组人促红细胞生成素的含量检测方法,包括如下步骤:
11.将重组人促红细胞生成素(rhepo)理化对照品与溶剂混合,制备不同浓度的对照品;
12.取重组人促红细胞生成素样品,制备待测品;
13.将所述不同浓度的对照品进行反相高效液相色谱法检测,建立浓度和峰面积的对应关系;
14.将所述待测品进行反相高效液相色谱法检测,将所得峰面积代入所述浓度和峰面积的对应关系,计算所述待测品中重组人促红细胞生成素的含量;
15.其中,所述反相高效液相色谱法采用的流动相包括流动相a和流动相b;
16.以体积百分比计,流动相a包括:65%~75%的水(h2o)和35%~25%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa);
17.以体积百分比计,流动相b包括:35%~45%的水(h2o)和65%~55%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa)。
18.在其中一个实施例中,所述反相高效液相色谱法采用的洗脱程序包括:
19.0~1min,保持流动相a的体积分数为100%,流动相b的体积分数为0%;
20.1min~25min,流动相a的体积分数由100%变化至0%,流动相b的体积分数由0%变化至100%;
21.25min~38min,保持流动相a的体积分数为0%,流动相b的体积分数为100%。
22.在其中一个实施例中,所述反相高效液相色谱法采用的洗脱程序还包括:
23.38min~43min,流动相a的体积分数由0%变化至100%,流动相b的体积分数由
100%变化至0%;
24.43min~55min,保持流动相a的体积分数为100%,流动相b的体积分数为0%。
25.在其中一个实施例中,所述反相高效液相色谱法采用的色谱柱以c4~c18烷基硅烷键合硅胶中的一种为填充剂。
26.在其中一个实施例中,所述反相高效液相色谱法采用的色谱柱以c4、c6、c8或c18烷基硅烷键合硅胶为填充剂。
27.在其中一个实施例中,所述反相高效液相色谱法的条件还包括:流速为0.9~1.1ml/min,温度25℃~35℃,紫外检测器波长为209nm~211nm。
28.在其中一个实施例中,所述反相高效液相色谱法的条件还包括:流速为1ml/min,温度28℃~32℃,紫外检测器波长为210nm。
29.在其中一个实施例中,检测结果的积分条件为:峰宽50,阈值200,积分区间为7min~30min。
30.在其中一个实施例中,重组人促红细胞生成素对应的峰的保留时间为19.3
±
0.2min。
31.在其中一个实施例中,所述浓度和峰面积的对应关系为y=74056.85x-108767.04,其中,x表示浓度,y表示所述浓度下重组人促红细胞生成素的峰面积。
32.上述重组人促红细胞生成素的含量检测方法,通过采用反相高效液相色谱法,并合理控制高效液相色谱法的检测条件,可以对待测样品中重组人促红细胞生成素进行准确测定,操作简便,无需进行如传统方法中复杂的抗体偶联等操作,与传统的生物学检测方法和免疫学检测方法相比,具有样品需求量少,检测成本低、耗时少,准确度高的优点,且还能检测成分复杂的样品如细胞培养上清,对重组人促红细胞生成素的工艺优化及生产的开展具有非常高的指导意义。
33.同时,上述重组人促红细胞生成素的含量检测方法经过了全面的验证,包括系统适用性、专属性、线性、准确度、精密度及中间精密度、定量限和检测限、耐用性等验证项目,具有较高的检测准确度,加标回收率在95.05%~100.66%之间,检测精密度高,这也使得该方法重复性好,定量限低至30μg/ml rhepo,使得细胞培养及下游纯化工艺中重组人促红细胞生成素含量测定结果更加准确可信。
附图说明
34.图1为线性验证空白溶液hplc典型图谱;
35.图2为线性验证20μg/ml供试品溶液hplc典型图谱;
36.图3为线性验证40μg/ml供试品溶液hplc典型图谱;
37.图4为线性验证80μg/ml供试品溶液hplc典型图谱;
38.图5为线性验证160μg/ml供试品溶液hplc典型图谱;
39.图6为线性验证320μg/ml供试品溶液hplc典型图谱;
40.图7为线性验证640μg/ml供试品溶液hplc典型图谱;
41.图8为系统适用性验证空白溶液hplc典型图谱;
42.图9为系统适用性验证系统适用性溶液hplc典型图谱;
43.图10为专属性验证空白溶液hplc图谱;
44.图11为专属性验证对照品hplc图谱;
45.图12为专属性验证供试品hplc图谱;
46.图13为精密度与中间精密度验证空白溶液hplc典型图谱;
47.图14为精密度与中间精密度验证供试品溶液hplc典型图谱;
48.图15为准确度验证空白溶液hplc典型图谱;
49.图16为准确度验证细胞上清hplc典型图谱;
50.图17为准确度验证30μg/ml加标样品hplc典型图谱;
51.图18为准确度验证330μg/ml加标样品hplc典型图谱;
52.图19为准确度验证630μg/ml加标样品hplc典型图谱;
53.图20为定量限与检测限验证验证空白溶液hplc典型图谱;
54.图21为检测限20μg/ml hplc典型图谱;
55.图22为定量限30μg/ml hplc典型图谱;
56.图23为200μg/ml对照品(28℃)hplc图谱;
57.图24为200μg/ml对照品(30℃)hplc图谱;
58.图25为200μg/ml对照品(32℃)hplc图谱;
59.图26为200μg/ml对照品(208nm)hplc图谱;
60.图27为200μg/ml对照品(210nm)hplc图谱;
61.图28为200μg/ml对照品(212nm)hplc图谱;
62.图29为200μg/ml对照品(0.8ml/min)hplc图谱;
63.图30为200μg/ml对照品(1.0ml/min)hplc图谱;
64.图31为200μg/ml对照品(1.2ml/min)hplc图谱。
具体实施方式
65.以下结合具体实施例对本发明的重组人促红细胞生成素的含量检测方法作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
66.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
67.本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
68.本发明中,涉及到数值区间,如无特别说明,上述数值区间内视为连续,且包括该范围的最小值及最大值,以及这种最小值与最大值之间的每一个值。进一步地,当范围是指整数时,包括该范围的最小值与最大值之间的每一个整数。此外,当提供多个范围描述特征或特性时,可以合并该范围。换言之,除非另有指明,否则本文中所公开之所有范围应理解为包括其中所归入的任何及所有的子范围。
69.本发明中涉及的百分比含量,如无特别说明,对于固液混合和固相-固相混合均指质量百分比,对于液相-液相混合指体积百分比。如无特别说明,溶液样品的溶剂均为水。
70.本发明中涉及的百分比浓度,如无特别说明,均指终浓度。所述终浓度,指添加成
分在添加该成分后的体系中的占比。
71.本发明中的温度参数,如无特别限定,既允许为恒温处理,也允许在一定温度区间内进行处理。所述的恒温处理允许温度在仪器控制的精度范围内进行波动。
72.本发明提供一种重组人促红细胞生成素的含量检测方法,包括如下步骤:
73.将重组人促红细胞生成素(rhepo)理化对照品与溶剂混合,制备不同浓度的对照品;
74.取重组人促红细胞生成素样品,制备待测品;
75.将所述不同浓度的对照品进行反相高效液相色谱法检测,建立浓度和峰面积的对应关系;
76.将所述待测品进行反相高效液相色谱法检测,将所得峰面积代入所述浓度和峰面积的对应关系,计算所述待测品中重组人促红细胞生成素的含量;
77.其中,所述反相高效液相色谱法采用的流动相包括流动相a和流动相b;
78.以体积百分比计,流动相a包括:65%~75%的水(h2o)和35%~25%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa);
79.以体积百分比计,流动相b包括:35%~45%的水(h2o)和65%~55%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa)。
80.可以理解地,所述待测样品可以为cho细胞培养上清及下游纯化工艺中的中间品。
81.在其中一些具体的示例中,所述溶剂为tris-hcl缓冲液。
82.在其中一些具体的示例中,以体积百分比计,流动相a包括:68%~72%的水(h2o)和32%~28%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa)。具体地,以体积百分比计,流动相a包括:70%的水(h2o)和30%的乙腈(acn);以及占水与乙腈总体积的0.1%的三氟乙酸(tfa)。
83.在其中一些具体的示例中,以体积百分比计,流动相b包括:38%~42%的水(h2o)和62%~58%的乙腈(acn);以及占水与乙腈总体积的0.05%~0.15%的三氟乙酸(tfa)。具体地,以体积百分比计,流动相b包括:40%的水(h2o)和60%的乙腈(acn);以及占水与乙腈总体积的0.1%的三氟乙酸(tfa)。
84.在其中一些具体的示例中,所述反相高效液相色谱法采用的洗脱程序包括:
85.0~1min,保持流动相a的体积分数为100%,流动相b的体积分数为0%;
86.1min~25min,流动相a的体积分数由100%变化至0%,流动相b的体积分数由0%变化至100%;
87.25min~38min,保持流动相a的体积分数为0%,流动相b的体积分数为100%。
88.进一步地,所述反相高效液相色谱法采用的洗脱程序还包括:
89.38min~43min,流动相a的体积分数由0%变化至100%,流动相b的体积分数由100%变化至0%;
90.43min~55min,保持流动相a的体积分数为100%,流动相b的体积分数为0%。
91.在其中一些具体的示例中,检测结果的积分条件为:峰宽50,阈值200,积分区间为7min~30min。
92.在其中一些具体的示例中,按面积归一化法计算重组人促红细胞生成素的峰面积。
93.在其中一些具体的示例中,所述反相高效液相色谱法采用的色谱柱以c4~c18烷基硅烷键合硅胶中的一种为填充剂。进一步地,所述反相高效液相色谱法采用的色谱柱以c4、c6、c8或c18烷基硅烷键合硅胶为填充剂。具体地,所述反相高效液相色谱法采用的色谱柱phenomenexc18柱。
94.在其中一些具体的示例中,所述反相高效液相色谱法采用的色谱柱参数包括:4.6mm
×
(150~250)mm,孔径12~30nm,粒径3.5~10μm。
95.在其中一些具体的示例中,所述反相高效液相色谱法的条件还包括:流速为0.9~1.1ml/min,温度25℃~35℃,紫外检测器波长为209nm~211nm。进一步地,所述反相高效液相色谱法的条件还包括:流速为1ml/min,温度28℃~32℃,紫外检测器波长为210nm。
96.在其中一些具体的示例中,重组人促红细胞生成素对应的峰的保留时间为19.3
±
0.2min。
97.在其中一些具体的示例中,所述浓度和峰面积的对应关系是指标准曲线。具体地,所述标准曲线为y=74056.85x-108767.04,其中,x表示浓度,y表示所述浓度下重组人促红细胞生成素的峰面积。。
98.以下为具体的实施例。
99.实施例1
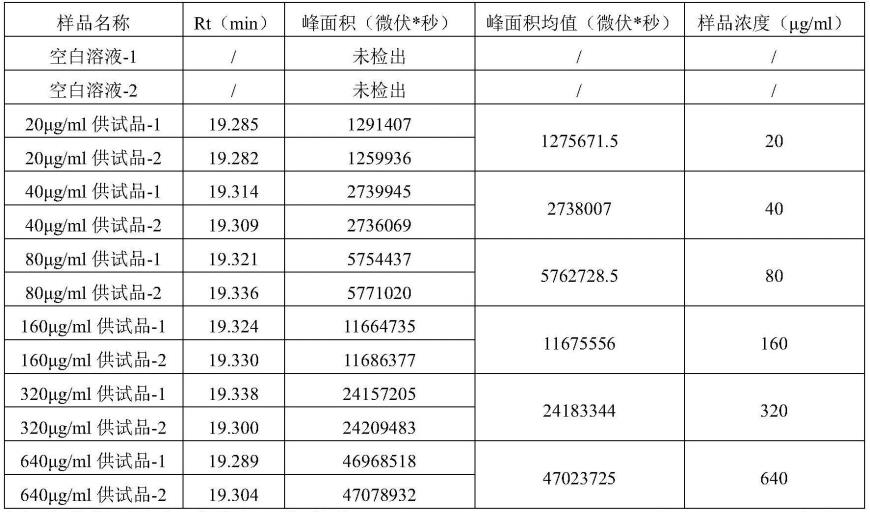
100.本实施例为一种重组人促红细胞生成素的含量检测方法,步骤如下:
101.(1)色谱柱:phenomenexc18柱,采用十八烷基硅烷键合硅胶为填充剂,4.6mm
×
250mm,cv=4.15ml,孔径30nm,粒径5μm,柱效≥25411。
102.(2)流动相:
103.流动相a(体积百分比):70%h2o+30%acn,0.1%tfa;
104.流动相b(体积百分比):40%h2o+60%acn,0.1%tfa。
105.(3)空白溶液:10mm tris-hcl缓冲液,ph7.00。
106.(4)rhepo理化对照品溶液:购自科兴生物制药股份有限公司,经过lowry法标定,蛋白浓度为3.77g/l,电泳纯度≥99%。
107.(5)待测品:cho细胞培养获得的含rhepo的培养上清液,经0.45μm膜过滤。
108.(6)高效液相色谱:
109.采用高效液相色谱系统(agilent 110series)进行样品检测,流速1ml/min,温度30℃,紫外检测器波长210nm,进样体积100μl,流动相洗脱梯度表如下表1所示:
110.表1检测方法溶液梯度表
111.时间(min)a(%)b(%)0100011000250100380100431000551000
112.液相图谱的积分条件为:峰宽50,阈值200,积分区间为7min~30min,按面积归一
化法计算rhepo的峰面积,目的蛋白峰保留时间19.3
±
0.2min。
113.(7)标准曲线构建:取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成20μg/ml、40μg/ml、80μg/ml、160μg/ml、320μg/ml、640μg/ml六个不同浓度的供试品溶液。将空白溶液和供试品按顺序各进样两次检测,以浓度为横坐标,扣除噪音后峰面积平均值为纵坐标,建立标准曲线,要求线性方程r2≥0.995。验证结果如下表2和图1~7所示:
114.表2线性验证结果
[0115][0116]
以上表数据建立标准曲线,方程为y=74056.85x-108767.04,r2=0.99971。由表2可知:以浓度为横坐标,扣除噪音后峰面积平均值为纵坐标,得到一条浓度与峰面积的线性关系式,其r2=0.99971,大于0.995,说明浓度与峰面积存在一定的线性关系,且线性度良好,线性验证合格。
[0117]
(8)检测结果:rhepo细胞培养上清浓度43.68μg/ml。
[0118]
实施例2
[0119]
本实施例对实施例1的重组人促红细胞生成素的含量检测方法的方法学验证。
[0120]
(1)系统适用性验证
[0121]
系统适用性验证是检查高效液相色谱系统、色谱柱是否符合检测要求,通过检查目的蛋白峰的保留时间(rt)、峰面积的相对标准偏差(rsd),确定检测结果是否准确可靠,是否适用于rhepo含量的检测。
[0122]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释至200μg/ml作为系统适用性溶液。空白溶液进样两次,系统适用性溶液连续进样四次,记录色谱图。要求空白溶液无干扰,系统适用性溶液连续进四次,主峰保留时间rsd≤1.0%,主峰面积rsd≤2.0%。验证结果如下表3和图8~9所示:
[0123]
表3系统适用性验证结果
[0124][0125][0126]
由表3和图8~9可以看出:空白溶液进样后在主峰的保留时间内无紫外吸收,说明空白溶液无干扰。系统适用性溶液主峰保留时间rsd及主峰面积rsd都在规定的范围内。因此,系统适用性验证合格,所使用的高效液相色谱系统、色谱柱符合检测要求,适用于rhepo含量的检测。
[0127]
(2)专属性验证
[0128]
专属性验证是指在其它成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力,对于rhepo细胞上清,成分较为复杂。rhepo纯化工序中都使用了tris-hcl缓冲液,因此使用10mm tris-hcl缓冲液对对照品进行稀释,用10mm tris-hcl缓冲液作为空白,验证其对rhepo含量测定有无干扰作用。
[0129]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释至40μg/ml作为对照品溶液。取rhepo细胞上清,作为供试品溶液。空白溶液、对照品溶液、供试品溶液各进一次,记录色谱图。要求空白溶液无干扰,供试品溶液主峰保留时间与对照品溶液主峰保留时间偏差应<1%。验证结果如下表4和图10~12所示:
[0130]
表4专属性验证结果
[0131][0132]
由表4和图10~12可以看出:空白溶液无干扰,供试品主峰保留时间与对照品溶液主峰保留时间偏差0.7%,保留时间基本一致。因此,其他成分(如杂质、降解产物、辅料等)、空白溶液存在的情况下,本检测方法也具有正确测定出被测物的能力,专属性验证合格。
[0133]
(3)精密度与中间精密度验证
[0134]
精密度与中间精密度验证是用于对仪器的性能检验,也确认分析人员的分析能力,保证检验结果准确可靠,重复性好。
[0135]
第一人取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释至200μg/ml作为供试品溶液,平行制备三份。空白溶液进样一次、供试品溶液每份各进一次,记录色谱图。要求空白溶液无干扰,第一人三份供试品溶液中,主峰保留时间rsd≤1.0%,主峰峰面积rsd≤5.0%。
[0136]
第二人取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释至200μg/ml作为
供试品溶液,平行制备三份。更换一台设备,空白溶液进样一次、供试品溶液每份各进一次,记录色谱图。要求空白溶液无干扰,第二人三份供试品溶液中,主峰保留时间rsd≤1.0%,主峰峰面积rsd≤5.0%;两人六份供试品溶液中主峰保留时间rsd≤1.0%,主峰峰面积rsd≤5.0%。验证结果如下表5和图13~14所示:
[0137]
表5精密度与中间精密度验证结果
[0138][0139][0140]
由表5和图13~14可以看出:第一人三份供试品溶液中,空白溶液无干扰,主峰保留时间rsd=0.032%,主峰峰面积rsd=1.59%。第二人三份供试品溶液中,空白溶液无干扰,主峰保留时间rsd=0.083%,主峰峰面积rsd=0.66%,两人六份样品中,主峰保留时间rsd=0.11%,主峰峰面积rsd=1.82%。第一人、第二人三份样品及两人六份样品,主峰保留时间及峰面积rsd均小于规定的合格范围。因此,仪器的性能及分析人员的分析能力均无问题,精密度与中间精密度验证合格。
[0141]
(5)准确度验证
[0142]
准确度验证是指本检测方法得到的测量值与真实值之间相近程度,用目的蛋白的回收率进行表示。
[0143]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成16μg/ml、616μg/ml、1216μg/ml三个浓度的对照品稀释液,取一份供试品溶液(rhepo细胞上清)按体积比1:1与对照品稀释液混合成三份不同浓度的加标样品,将上述三份加标样品分别进样三次,供试品进样两次,计算浓度和加标回收率。要求空白溶液无干扰,各浓度下主峰平均加标回收率在90%-110%之间,三组样品各自加标回收率rsd≤5%,9个样品加标回收率rsd≤5%。验证结果如下表6和图15~19所示:
[0144]
表6准确度验证结果
[0145][0146]
由表6和图15~19可知:各浓度下的平均加标回收率为95.05%~100.66%,三组样品各自加标回收率rsd为0.10%~2.24%,9个样品加标回收率rsd为2.79%,所得的加标回收率及rsd均在规定的合格范围内,本检测方法得到的测量值与真实值之间相近程度高,相对标准偏差较小,因此,准确度验证合格。
[0147]
(6)定量限与检测限验证
[0148]
定量限度是指在合适的准确性和精密度下,能够定量测定样品中被分析物的最低量。检测限是指样品中的被分析物能够被检测到的最低量,但不一定要准确定量。
[0149]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成20μg/ml、30μg/ml,分别作为检测限样品和定量限样品。取空白溶液进样两次,检测限样品进样两次,定量限样品进样三次。要求检测限样品能检出目的蛋白,峰面积均值与噪音峰面积均值的比值(信噪比)≥3,定量限样品峰面积均值与噪音峰面积均值的比值≥5,定量限样品峰面积rsd≤10%,主峰留时间rsd≤2%。验证结果如下表7和图20~22所示:
[0150]
表7定量限与检测限验证结果
[0151][0152]
由表7和图20~21可以看出:当样品浓度为20μg/ml时,由于空白溶液在主峰的保留时间内无紫外吸收,信噪比为41,远大于3,从图21可以看出此时主峰的响应值已较低,若继续降低浓度也能检出主峰但峰型会较差,且20μg/ml的检测限可以满足行业一般的工艺要求,所以将本rhepo含量检测方法的检测限定为20μg/ml。
[0153]
当样品浓度为30μg/ml时,信噪比为73,远大于5,连续进样三次,主峰峰面积rsd=0.44%,主峰保留时间rsd=0.06%,均小于规定的合格范围,加上前期在线性及准确度验证中已经证明30μg/ml具有良好的线性度及准确度,所以将本rhepo含量检测方法的定量限
定为30μg/ml可以满足行业一般的工艺要求。
[0154]
(7)耐用性验证
[0155]
7.1不同柱温下检测结果对比
[0156]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成200μg/ml,保持其他条件不变,分别在28℃、30℃、32℃条件下检测,观察检测结果,如下表8和图23~25所示:
[0157]
表8不同柱温对rhepo含量检测的影响
[0158][0159]
从表8和图23~25可以看出:其他条件不变,当柱温30℃
±
2℃范围内波动时,本检测方法具有一定的稳定性,对rhepo含量检测不会造成影响。
[0160]
7.2不同波长下检测结果对比
[0161]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成200μg/ml,保持其他条件不变,分别在208nm、210nm、210nm条件下检测,观察检测结果,如下表9和图26~28所示:
[0162]
表9不同波长对rhepo含量检测的影响
[0163][0164]
从表9可以看出:其他条件不变,当检测波长为208nm时,此时测得的rhepo含量相比于正常条件下偏差了16.74%,造成偏差的原因可能为:本检测方法使用的有机溶剂为乙腈,乙腈的紫外吸收截止波长为190~210nm,当波长<210nm时可能存在部分乙腈被吸收,从而导致目的蛋白浓度测量值偏高。当检测波长为212nm时,此时测得的rhepo含量相比于正常条件下偏差了-12.54%,造成偏差的原因可能为:可能存在部分目的蛋白没有被紫外吸收,导致目的蛋白浓度测量值偏低。
[0165]
综上所述,本rhepo含量检测方法对检测波长较为敏感,波长变动会对检测结果造成较大的影响,因此在检测过程中应按照检测波长为210nm执行。
[0166]
7.3不同流速下检测结果对比
[0167]
取rhepo理化对照品溶液,使用10mm tris-hcl缓冲液稀释成200μg/ml,保持其他条件不变,分别在0.8ml/min、1.0ml/min、1.2ml/min条件下检测,观察检测结果,如下表10和图29~31所示:
[0168]
表10不同流速对rhepo含量检测的影响
[0169][0170]
从表10可以看出:其他条件不变,当检测流速为0.8ml/min时,此时测得的rhepo含量相比于正常条件下偏差了25.20%,造成偏差的原因可能为:流速降低之后流动相的传质速度相对变慢,主峰保留时间滞后,峰型较宽(体积大、浓度低),加上本验证所使用的hplc的检测器为浓度型检测器,峰面积与流动相流速成反比,因此造成了峰面积相对于正常条件下检测值偏高,从而测得的浓度偏大。当检测流速为1.2ml/min时,流动相的传质速度相对变快,主峰保留时间提前,峰型尖锐其窄小(体积小、浓度高),可能导致积分出来的峰面积偏小,所测得的浓度相对于正常条件偏低。
[0171]
综上所述,本rhepo含量检测方法对检测流速较为敏感,流速变动会对检测结果造成较大的影响,因此在检测过程中应严格按照检测流速为1.0ml/min执行。
[0172]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0173]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1