表面增强拉曼散射及分光光度双模式检测甲基汞及赭曲霉毒素A的方法
表面增强拉曼散射及分光光度双模式检测甲基汞及赭曲霉毒素a的方法
技术领域
1.本发明属于化学分析检测技术领域,具体为一种表面增强拉曼散射及分光光度双模式检测甲基汞及赭曲霉毒素a的方法。
背景技术:
2.汞及其化合物广泛存在于自然界中,汞的毒性与其存在的形态密切相关,有机汞的毒性大于无机汞,在有机汞中甲基汞(mehg)的毒性最大。无机汞在生物体烷基化的作用下可生成甲基汞,汞及甲基汞被动植物吸收后通过食物链的富集作用进入人体,其富集倍数可达106~107。食品安全国家标准gb 5009.17-2017《食品中总汞及有机汞的测定》中甲基汞的测定方法有高效液相色谱-原子荧光联用法、高效液相色谱-电感耦合等离子体质谱法。赭曲霉素a(ota)是自然界中最普遍、最有毒的真菌毒素之一,是曲霉和青霉菌产生的次级代谢产物。ota作为一种普遍存在的污染物,广泛存在于咖啡、大米、花生、肉、酒、玉米等多种食品中。此外,ota最终可以通过食物链的积累进入人体,威胁人体健康。由于ota具有严重的致癌性、肝毒性和肾毒性,国际癌症研究机构(iarc)和世界卫生组织(who)将ota列为人类2b类致癌物。食品中ota的水平一直受到严格的调控。参照欧盟和中国标准,葡萄酒中ota的最高允许量为2μg/kg,谷物和咖啡制品中ota的最高允许量为5μg/kg。目前,已经发展了薄层色谱法(tlc)、酶联免疫吸附法(elisa)、高效液相色谱法(hplc)和质谱(ms)等多种ota定量分析方法。虽然这些方法在灵敏度和可靠性方面取得了进展,但由于操作复杂、设备庞大、成本高、耗时长,限制了它们在快速现场检测中的实际应用。因此,开发一种简单、方便、灵敏的ota监测方法具有重要意义。
3.表面增强拉曼光谱(surface-enhanced raman spectroscopy,sers)通过金、银等贵金属材料大幅增强目标物质的拉曼信号,实现痕量物质的检测,其除了具有指纹谱图特征外,还具有样品前处理简单、检测速度快、所需样品少等优点,进而在食品安全检测领域居于独特的地位。但由于目前拉曼增强剂制备方法不多,能够产生拉曼信号的靶标有限,sers应用并不广泛。分光光度法由于简便、快速、不需要复杂仪器设备,被广泛应用,但易受到共存物质的干扰。将分光光度法和sers相结合,可以方便地从复杂的矩阵中筛选目标物,降低了分析检测中出现假阳性或假阴性的概率。
技术实现要素:
4.针对现有技术的不足,本发明提供了一种表面增强拉曼散射及分光光度双模式检测甲基汞及赭曲霉毒素a的方法,该方法基于巯基-β-环糊精基碳点(sh-β-cds)改性金纳米具有的模拟酶及拉曼增加的双重活性,在双氧水存在下氧化无色隐性孔雀石绿为有可见光吸收及表面增强拉曼光谱活性的孔雀石绿,而甲基汞的加入增强了金纳米的过氧化酶活性,由于金银汞齐的形成,同时也增加了拉曼活性,当赭曲霉毒素a存在时,能与甲基汞相互作用而抑制纳米酶活性,从而减弱了隐性孔雀石绿的氧化,导致吸光度及sers信号降低;基
于甲基汞浓度与底物孔雀石绿的吸光度及sers增加的呈线性关系,同时基于赭曲霉毒素a浓度与底物孔雀石绿的吸光度及sers降低的线性关系,建立高灵敏、选择性强的甲基汞及赭曲霉毒素a的双模式紫外及sers检测方法,甲基汞及赭曲霉毒素a的sers定量限分别为0.34
µ
g/l及0.25
µ
g/l,分光光度法测定的定量限分别为1.25
µ
g/l及0.75
µ
g/l;本方法具有操作简单、灵敏度高、快速等特点。
5.本发明表面增强拉曼散射及分光光度双模式检测甲基汞及赭曲霉毒素a的方法如下:(1)以巯基-β-环糊精基碳点(sh-β-cds)、柠檬酸钠为还原剂及稳定剂,制备sh-β-cds改性金纳米aunps;(2)在甲基汞标准溶液中加入sh-β-cds改性金纳米aunps溶液,室温反应3-5min,然后加入隐性孔雀石绿(lmg)溶液及h2o2,生成绿色孔雀石绿;采用紫外-可见分光光度法检测反应产物,在588nm波长处测定吸光度,以甲基汞浓度为横坐标,吸光度为纵坐标,确定甲基汞浓度与吸光度的线性关系;采用表面增强拉曼散射光谱检测反应产物,根据孔雀石绿分子结构和拉曼峰位归属,确定794cm-1
处的特征峰作为表面增强拉曼散射光谱检测甲基汞的判别依据,并确定甲基汞浓度与特征峰的峰面积的线性关系;先在30μg/l甲基汞溶液中加入赭曲霉毒素a标准溶液反应5-10mim后,再加入sh-β-cds改性金纳米aunps溶液,然后加入隐性孔雀石绿溶液及h2o2,生成绿色孔雀石绿;采用紫外-可见分光光度法检测反应产物,在588nm波长处测定吸光度,以赭曲霉毒素a浓度为横坐标,吸光度为纵坐标,确定赭曲霉毒素a浓度与吸光度的线性关系;采用表面增强拉曼散射光谱检测反应产物,根据孔雀石绿分子结构和拉曼峰位归属,确定794cm-1
处的特征峰作为表面增强拉曼散射光谱检测赭曲霉毒素a的判别依据,并确定赭曲霉毒素a浓度与特征峰的峰面积的线性关系;(3)取待测甲基汞的样品液与sh-β-cds改性金纳米aunps溶液混合反应后,加入隐性孔雀石绿、h2o2反应,分别进行紫外-可见分光光度及表面增强拉曼光谱检测,根据吸光度和特征峰的峰面积计算待测样品液中甲基汞浓度;取待测赭曲霉毒素a的样品液与甲基汞溶液混合反应后,再加入sh-β-cds改性金纳米aunps、隐性孔雀石绿、h2o2反应,分别进行紫外-可见分光光度及表面增强拉曼光谱检测,根据吸光度和特征峰的峰面积计算待测样品液中赭曲霉毒素a浓度。
6.所述sh-β-cds改性金纳米aunps的制备方法如下:(1)称取0.05-0.1g巯基-β-环糊精、0.03-0.05g去甲肾上腺素、2.0-2.5g柠檬酸、0.05-0.1g乙二胺溶于40-60ml超纯水中,超声混合均匀,将溶液转移至聚四氟乙烯内衬水热反应罐中,置于微波消解仪中,于180℃消解1h,反应完成后自然冷却至室温,得棕色溶液;将棕色溶液用0.22μm滤膜除去大颗粒杂质,再经高速离心,上清液真空干燥,得到巯基-β-环糊精碳点sh-β-cds;(2)将1-1.5mg巯基-β-环糊精碳点sh-β-cds、20-25mg柠檬酸钠溶于40-60ml超纯水中,再加入haucl4,85-95℃加热搅拌25-30min,即得sh-β-cds改性金纳米aunps,其中haucl4在混合液中的浓度为10-15mg/ml。
7.表面增强拉曼散射光谱检测中甲基汞标准溶液的浓度为0.34-50.00μg/l,赭曲霉毒素a标准溶液浓度为0.25-41.78μg/l;紫外-可见分光光度法检测中甲基汞标准溶液的浓
度为1.25-46.87μg/l,赭曲霉毒素a标准溶液的浓度为0.75-68.75μg/l;sh-β-cds改性金纳米aunps溶液浓度为0.1mg/ml,添加量为45-55
µ
l;隐性孔雀石绿溶液浓度为5mmol/l,添加量为80-120
µ
l;h2o2的浓度为50mmol/l,添加量为45-55
µ
l。
8.所述的拉曼光谱检测是使用便携式拉曼检测仪,在785nm激发光、激光功率500mw条件下扫描10s。
9.所述的离心是在8000-10000 r/min下处理10-15min。
10.本发明的优点和技术效果:1、本发明采用巯基-β-环糊精基碳点(sh-β-cds)改性金纳米具有的模拟酶及拉曼增加的双重活性,在双氧水存在下氧化无色隐性孔雀石绿为有可见吸收及表面增强拉曼光谱活性的孔雀石绿,而甲基汞的加入增强了改性金纳米的过氧化酶活性,一方面sh-β-cds改性金纳米aunps能与甲基汞形成金银汞齐合金,同时,sh-β-cds中的sh-与au或hg形成s-au或s-hg键,增强改性金纳米aunps的过氧酶活性,同时也增加了拉曼活性,当赭曲霉毒素a存在时,能与甲基汞相互作用而抑制改性金纳米aunps活性,从而减弱了隐性孔雀石绿的氧化,导致吸光度及sers信号降低,基于此建立了甲基汞及赭曲霉毒素a的分光光度及sers双模式检测方法;2、本发明建立的甲基汞及赭曲霉毒素a的分光光度法及sers检测方法,具有高的检测灵敏度,甲基汞及赭曲霉毒素a的sers的定量限分别为0.34
µ
g/l及0.25
µ
g/l,分光光度测定的定量限分别为1.25
µ
g/l及0.75
µ
g/l,而共存的hg
2+
、其他真菌毒素不干扰测定,方法具有好的选择性;3、本发明方法利用先将赭曲霉毒素a与甲基汞反应,利用ota中氧与hg形成配位键,后续sh-β-cds改性金纳米材料能在与甲基汞形成au-hg合金时,反应受到抑制,导致aunps纳米酶活性降低,从而引起体系吸光度及sers信号降低,将分光光度法和sers相结合,可以方便地从复杂的矩阵中筛选目标物,降低了分析检测中出现假阳性或假阴性的概率。
附图说明
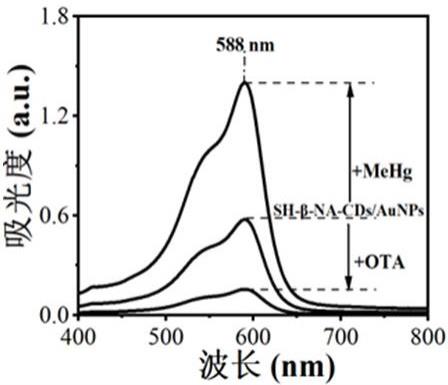
11.图1为本发明实施例1中sh-β-cds/aunps+lmg+h2o2、sh-β-cds/aunps+mehg+lmg+h2o2及sh-β-cds/aunps+mehg+lmg+h2o2+ota紫外吸收光谱图;图2为本发明实施例1中sh-β-cds/aunps+lmg+h2o2、sh-β-cds/aunps+mehg+lmg+h2o2及sh-β-cds/aunps+mehg+lmg+h2o2+ota的sers图;图3为本发明实施例1中不同浓度甲基汞标准液的sers图及线性关系图;图4为本发明实施例1中不同浓度赭曲霉毒素a标准液的sers图及线性关系图;图5为本发明实施例1中不同浓度甲基汞标准液的紫外-可见光谱图及线性关系图;图6为本发明实施例1中不同浓度赭曲霉毒素a标准液的紫外-可见光谱图及线性关系图;图7为不同物质对本发明检测体系的干扰实验结果;图8为不同物质对本发明sers检测体系的干扰实验结果;图9为不同真菌毒素及其他物质对本发明检测体系的干扰实验结果;
图10为不同真菌毒素及其他物质对本发明sers检测体系的干扰实验结果。
具体实施方式
12.下面将结合具体的实施例对本发明的技术方案作进一步详细地描述说明,但本发明的保护范围并不仅限于此。
13.实施例1:饲料中ota及食用菌中mehg的测定1、巯基-β-环糊精基碳点改性金纳米aunps的制备(1)称取0.05g巯基-β-环糊精、0.03g去甲肾上腺素、2.0g柠檬酸、0.05g乙二胺溶于40ml超纯水中,超声混合均匀,将溶液转移至聚四氟乙烯内衬水热反应罐中,置于微波消解仪中,于180℃消解1h,反应完成后自然冷却至室温,得棕色溶液;将棕色溶液用0.22μm滤膜除去大颗粒杂质,再经高速离心,上清液真空干燥,得到巯基-β-环糊精基碳点sh-β-cds;(2)将1mgsh-β-cds、20mg柠檬酸钠溶于40ml超纯水中,再加入haucl4,haucl4在混合液中的浓度为10mg/ml,90℃加热搅拌25min,即得sh-β-cds改性金纳米aunps(sh-β-cds/aunps);在588nm波长处测定吸光度,由图1可以看出,sh-β-cds改性金纳米aunps在mehg作用下能使可见光谱吸收信号增强,当ota存在时,ota能与甲基汞相互作用而抑制sh-β-cds改性金纳米aunps可见光谱吸收信号;使用便携拉曼仪在794cm1处进行拉曼光谱检测,由图2可以看出,sh-β-cds改性金纳米aunps在mehg作用下能使sers信号增强,当ota存在时,ota能与甲基汞相互作用而抑制sh-β-cds改性金纳米aunps拟pod活性,sers信号降低;2、mehg及ota标准品的sers分析甲基汞检测中,在样品瓶中加入甲基汞标准溶液200μl、0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,涡旋混合30秒,反应5分钟,接着加入50mmol/l的h2o250μl、5mmol/l的隐性孔雀石绿溶液100μl,甲基汞标准溶液的浓度为0.34-50.00μg/l,室温下反应10分钟,使用便携拉曼仪对混合液进行sers检测,在785nm激发光、激光功率500mw条件下扫描10s;赭曲霉毒素a测定中,在样品瓶中加入240μl浓度为0.5mg/l 的甲基汞溶液(即30μg/l)、赭曲霉毒素a标准溶液200μl,赭曲霉毒素a标准溶液的浓度为0.25-41.78μg/l,反应5分钟后再加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,然后加入50mmol/l的h2o250μl、5mmol/l的隐性孔雀石绿溶液100μl,用水稀释至4ml,室温下反应10分钟,使用便携拉曼仪对混合液进行sers检测,在785nm激发光、激光功率500mw条件下扫描10s;结果见图3、4,从甲基汞的sers谱图观察到794cm-1
、1172cm-1
、1219cm-1
、1394cm-1
和1615cm-1
处有sers特征峰,据孔雀石绿分子结构和拉曼峰位归属,确定794cm-1
处的特征峰作为sers光谱检测mehg及ota的检测依据;mehg及ota的sers谱中特征峰峰强随标准溶液浓度(0.34、1.67、3.33、6.67、13.33、20、26.67、33.33和50μg/l)及(0.25、1.67、8.33、16.67、25和41.78μg/l)而变化,如图3、4所示,mehg及ota浓度与特征峰峰面积的在797cm-1
处线性回归方程为:i= 13797c+40216,r2=0.988;i= 2240c+10756,r2=0.976;且均在浓度低至0.34
µ
g/l及0.25
µ
g/l时仍有明显的拉曼谱峰;所以,本方法对mehg及ota标准溶液的检出浓度为0.34
µ
g/l及0.25
µ
g/l;3、mehg及ota标准品的比色分析
mehg测定时,在样品瓶中加入甲基汞标准溶液200μl、0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,涡旋混合30秒,反应5分钟,接着加入50mmol/l的h2o250μl、50mmol/l隐性孔雀石绿溶液100μl,甲基汞标准溶液的浓度为1.25-46.87μg/l,室温下反应10分钟,在588nm波长处测定吸光度a;赭曲霉毒素a测定,在样品瓶中加入240μl浓度为0.5mg/l 的甲基汞溶液、赭曲霉毒素a标准溶液200μl,赭曲霉毒素a标准溶液浓度为0.75-68.75μg/l,反应5分钟后再加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,然后接着加入50mmol/lh2o250μl、5mmol/l隐性孔雀石绿溶液100μl,用水稀释至4ml,室温下反应10分钟,588nm波长处测定吸光度a;结果见图5、6所示,mehg及ota浓度与吸光度线性回归方程分别为:a= 0.031c+0.155,r2=0.993;a=0.025c+1.843,r2=0.996;且均在浓度低至1.25
µ
g/l及0.75
µ
g/l时仍有明显的吸收峰;所以,本方法对mehg及ota标准溶液的检出浓度为1.25
µ
g/l及0.75
µ
g/l。
14.4、食用菌中mehg含量的测定(1)样品前处理方法:称取研磨细的食用菌样品0.2g于萃取用玻璃管中,放入磁性搅拌子,加入10ml萃取液(0.07mol/l的hcl),用微波萃取仪在55℃、压力15mpa、功率110w下萃取15min;萃取完成后放入冰箱中静置5min,将上清液转移至离心管中,14 000r/min、4℃下离心5 min,吸取上清液,得到样品提取液;(2)样品中mehg测定:在样品提取液200μl中加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,涡旋混合30秒,反应5分钟,接着加入50mmol/l的h2o250μl、5mmol/l隐性孔雀石绿溶液100μl,室温下反应10分钟,在588nm波长处测定吸光度a,代入步骤3的回归方程,结果未检出;在785nm激发光、激光功率500mw条件下扫描10s,使用便携拉曼仪在794cm1处进行拉曼光谱检测,将采集得到的拉曼信号强度代入步骤2的回归方程,计算食用菌中甲基汞浓度为未检出。
15.5、饲料中ota含量的测定(1)样品前处理方法:称取5g猫科猪饲料,加入25ml溶剂(乙腈∶甲醇∶水=50∶40∶10,体积比),涡旋处理60s,然后振荡处理10min;在3000r/min转速下离心10min;取出上层溶液,重复提取2次,合并上层溶液,置入旋转蒸发仪,旋蒸至体积为1-2ml,得到样品提取液;(2)取样品提取液200μl,加入240μl浓度为0.5mg/l 的甲基汞溶液,反应5分钟后再加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,接着加入50mmol/l的h2o250μl、5mmol/l的lmg溶液100μl,室温下反应10分钟,在588nm处测定吸光度a,代入步骤3的回归方程,计算获得饲料中ota浓度为0.79μg/kg;同时在785nm激发光、激光功率500mw条件下扫描时间10s,使用便携拉曼仪在794cm-1
处进行拉曼光谱检测,将采集得到的拉曼信号强度代入步骤2的回归方程,计算获得饲料中ota浓度为0.82μg/kg;实施例2:咖啡中ota及草鱼中mehg的测定1、巯基-β-环糊精碳点改性金纳米的制备(1)称取0.08g巯基-β-环糊精、0.04g去甲肾上腺素、2.2g柠檬酸、0.07g乙二胺溶于50ml超纯水中,超声混合均匀,将溶液转移至聚四氟乙烯内衬水热反应罐中,置于微波消解仪中,于180℃消解1h,反应完成后自然冷却至室温,得棕色溶液;将棕色溶液用0.22μm滤膜除去大颗粒杂质,再经高速离心,上清液真空干燥,得到巯基-β-环糊精基碳点sh-β-cds;
(2)将1.2mgsh-β-cds、22mg柠檬酸钠溶于50ml超纯水中,再加入haucl4,haucl4在混合液中的浓度为12mg/ml,90℃加热搅拌30min,即得sh-β-cds改性金纳米aunps;2、mehg标准溶液的可见光谱及sers线性回归方程确定同实施例1;3、ota标准溶液的可见光谱及sers线性回归方程确定同实施例1;4、草鱼中mehg含量的测定(1)草鱼样品提取:称取10g(精确至0.001g)鲤鱼、5mol/l盐酸溶液50ml于100ml带盖塑料离心管中,室温下超声水浴提取60分钟,4℃、8000r/min下离心15分钟,取出上清液,在冷水浴中缓慢滴加氨水溶液(氨水与水体积比1:1混合制得),调节样品溶液的ph为6,加水至50ml,得样品提取液;(2)样品萃取:用20ml二氯甲烷分两次振荡萃取样品提取液,每次振荡10分钟,静置10分钟,收集合并二氯甲烷萃取液至50ml比色管中,用刻度吸管精确加入2ml反萃取溶液(含1%半胱氨酸、0.8%乙酸铵的水溶液)进行萃取,振荡5分钟后静置10分钟,吸取上层水溶液,得到待测样品液;(3)样品中mehg测定:在待测样品液200μl中加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,反应5分钟,接着加入50mmol/l的h2o250μl、5mmol/l的lmg溶液100μl,室温下反应10分钟,在588nm处测定吸光度a,代入步骤3的回归方程,计算获得草鱼中甲基汞浓度为1.19μg/kg;在785nm激发光、激光功率500mw条件下扫描10s,使用便携拉曼仪在794 cm-1
处进行拉曼光谱检测,将采集得到的拉曼信号强度代入步骤2的回归方程,计算草鱼中mehg浓度为1.23μg/kg;5、咖啡豆中ota含量的测定(1)样品前处理方法:取5g磨碎烘焙咖啡豆,加入25ml溶剂(乙腈∶甲醇∶水=50∶40∶10,v/v),涡旋处理60s,然后振荡处理10min;在3000r/min转速下离心10min;取出上层溶液,重复提取2次,合并上层溶液,置入旋转蒸发仪,旋蒸至体积为1-2ml,得到样品提取液;(2)样品中ota测定:取待测样品液200μl加入240μl浓度为0.5mg/l 的甲基汞溶液,反应5分钟后再加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,接着加入50mmol/l的h2o250μl、5mmol/l的lmg溶液100μl,室温下反应10分钟,在588nm处测定吸光度a,代入步骤3的回归方程,咖啡豆中ota未检出;在785nm激发光、激光功率500mw条件下扫描10s,使用便携拉曼仪在797cm-1
处进行拉曼光谱检测,将采集得到的拉曼信号强度代入步骤3的回归方程,咖啡豆中ota未检出。
16.实施例3:虾中mehg以及红酒中ota的测定(1)称取0.1g巯基-β-环糊精、0.05g去甲肾上腺素、2.5g柠檬酸、0.1g乙二胺溶于60ml超纯水中,超声混合均匀,将溶液转移至聚四氟乙烯内衬水热反应罐中,置于微波消解仪中,于180℃消解1h,反应完成后自然冷却至室温,得棕色溶液;将棕色溶液用0.22μm滤膜除去大颗粒杂质,再经高速离心,上清液真空干燥,得到巯基-β-环糊精基碳点sh-β-cds;(2)将1.5mg巯基-β-环糊精基碳点sh-β-cds、25mg柠檬酸钠溶于60ml超纯水中,再加入haucl4,haucl4在混合液中的浓度为15mg/ml,90℃加热搅拌30min,即得sh-β-cds改性金纳米aunps;2、mehg标准溶液的可见光谱及sers线性回归方程确定同实施例1;3、ota标准溶液的可见光谱及sers线性回归方程确定同实施例1;
4、虾样品中甲基汞含量的测定(1)虾样品的制备方法同实施例2草鱼样品;(2)样品中mehg测定:同实施例2,虾中mehg分光光度法测定浓度为0.90μg/kg,sers测定浓度为0.98μg/kg;5、红酒中ota含量的测定(1)样品前处理方法:准确称取红酒20.0g,准确到0.1g,置于25ml容量瓶中,加入提取液定容至刻度,混匀,经玻璃纤维滤纸过滤,收集滤液为样品提取液,其中提取液为将150.0g nacl、20.0gnahco3溶于约950ml纯净水中,加水定容至1l制得;(2)取样品提取液200μl,加入240μl浓度为0.5mg/l 甲基汞溶液,反应5分钟后再加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,接着加入50mmol/l的h2o250μl、5mmol/l的lmg溶液100μl,室温下反应10分钟,用水稀释至4ml,在588nm处测定吸光度a,代入步骤3的回归方程,咖啡豆中ota未检出;在785nm激发光、激光功率500mw条件下扫描10s,使用便携拉曼仪在794cm-1
处进行拉曼光谱检测,拉曼信号强度代入步骤2的回归方程,红酒样品中ota未检出。
17.上述实施例中检测样品的回收率与精密度实验:在实施例1-3检测样品中分别添加2个不同浓度的甲基汞标准溶液及赭曲霉毒素a标准溶液;每个浓度平行测定3次,计算加标回收率,并计算出相对标准偏差rsd,结果见表1及表2;测得mehg的加标回收率在92.1%~102.3%,rsd在3.29%~5.78%,ota的加标回收率在91.7%~98.2%,rsd在3.67%~5.81%,本方法有好的的准确性和精密度。
18.表1样品甲基汞加标回收率及rsd(n = 3)表2 样品中ota加标回收率及rsd(n = 3)
。
19.方法特异性考察:往1ml离心管中分别添加500μl浓度为0.1g/l的na
+
、k
+
、ca
2+
、mg
2+
、cr
3+
、mn
2+
、fe
2+
、fe
3+
、co
2+
、ni
2+
、cu
2+
、zn
2+
、ag
+
、cd
2+
、pb
2+
、hg
2+
(以上均为氯盐,体系中浓度折算为12.5mg/l)、240μl浓度0.5mg/l 的mehg(即30μg/l),加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,混匀,放置5min后加入100μl lmg溶液(5mmol/l)和50μl的h2o2(50mmol/l),用水稀释至4ml,室温放置10min后,在588nm下检测吸光度以及794 cm-1
处的sers强度变化;从图7及图8中可以看出,甲基汞检测的紫外-可见分光光度及sers体系有较好的选择特异性,其他物质对改性金银纳米没有增强活性作用。
20.一般情况下,食品中ota 与afb1、afb2、afg1和afg2等共存,将ota替换为其他物质,检测其他物质在上述检测体系中是否对甲基汞增强金纳米的过氧化酶活性有影响,往装有240μl浓度0.5mg/l mehg溶液的15ml离心管中分别添加500μl浓度0.1g/l的黄曲霉素b1(afb1)、玉米赤霉烯酮(zen)、伏马菌素b1(fb1)、脱氧雪腐镰刀菌烯醇(niv)、展青霉素(ptl)(体系中浓度折算为100μg/l)、250μl浓度1mg/l 的赭曲霉素a(ota)(即65μg/l),反应5min后加入0.1mg/ml的sh-β-cds改性金纳米aunps溶液50
µ
l,然后加入100μl lmg溶液(5mmol/l)和50μl的h2o2(50mmol/l),用水稀释至4ml,空白组是不添加真菌毒素,室温放置10min后,检测588nm紫外吸光度以及794cm-1
处的sers强度变化;从图9及图10中可以看出,在添加ota的检测体系中,赭曲霉毒素a能与甲基汞相互作用而抑制改性金纳米aunps活性,从而减弱了隐性孔雀石绿的氧化,导致吸光度及sers信号降低;其他真菌毒素没有抑制活性,因此检测体系中共存真菌毒素对ota检测没有干扰。
21.本发明建立的ota及甲基汞测定法具有处理步骤少,所用时间短,处理成本低,操作简便,不需要大型仪器设备,在实际检测中具有较强优势。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1