一种氮掺杂碳纳米SnO2复合材料及其制备方法和应用与流程
本发明属于材料技术领域,具体地,涉及一种氮掺杂碳纳米sno2复合材料及其制备方法和应用。
背景技术:
sno2纳米材料由于其材料成本低廉、制备方法简单、无毒无污染、形貌及微观结构易控、比容量高、热稳定性和化学稳定性高等优势,在能量存储、传感、催化等领域有着广泛的应用。但sno2本身低电导率的特性严重制约了其应用性能的提高,且纳米化的sno2由于较高的表面能极易发生团聚使其在应用过程的活性迅速下降。解决上述问题是提高sno2纳米材料应用性能的关键。
将纳米sno2与具有较高电导率特性的材料复合是同时解决上述问题的经典方法之一。较高的电导率有助于电子的快速迁移,同时复合材料的引入能有效抑制sno2纳米颗粒的团聚。目前已报道的复合材料包括掺杂型碳、石墨烯、导电聚合物、mxene等。其中氮掺杂碳因原料来源广、制备方法简单,且具有优秀的电化学性能而成为纳米sno2复合材料的最佳选择。例如,ma等人以聚丙烯腈和纳米sno2粉末为原料,通过静电纺丝和碳化过程制备了氧空位纳米sno2/氮掺杂碳纳米纤维复合材料(sno2-x/c),其电子迁移率达到225msm-1远高于sno2的0.00547msm-1(dingtaoma,yongliangli,hongweimi,shanluo,peixinzhang,zhiqunlin,jianqingli,andhanzhang,angew.chem.int.ed.2018,57,8901-8905)。
然而,该些现有的氧空位纳米sno2/氮掺杂碳纳米纤维复合材料的性能仍然无法有效满足现有的需求,且制备方法复杂。因此,如何快速高效的制备出sno2尺寸均一、均匀分散的氮掺杂碳纳米sno2复合材料,有效利用纳米sno2和氮掺杂碳基质之间的协同作用共同提高其应用性能仍面临巨大挑战。
技术实现要素:
针对上述挑战,本发明的目的在于提供一种氮掺杂碳纳米sno2复合材料及其制备方法和应用。本发明中的氮掺杂碳纳米sno2复合材料仅通过一步碳化锡-氮配位前驱体即可制备出sno2颗粒尺寸均一、均匀分散的氮掺杂碳纳米sno2复合材料。
本发明提供了一种氮掺杂碳纳米sno2复合材料的制备方法,其包括下述步骤:
(1)将含sn2+的溶液和聚4-乙烯基吡啶溶液混合,搅拌形成配合物沉淀,将所述配合物沉淀除去溶剂即得碳化前驱体;
所述聚4-乙烯基吡啶溶液中,溶剂为乙醇和/或水;
(2)将所述碳化物前驱体经碳化,即得所述氮掺杂碳纳米sno2复合材料。
步骤(1)中,所述含sn2+的溶液可为本领域常规的含sn2+的溶液,例如,将含sn2+的溶质和溶剂混合溶解,即得所述含sn2+的溶液。
其中,所述含sn2+的溶质可为氯化亚锡。所述氯化亚锡的存在形式可为无水氯化亚锡、二水合氯化亚锡或五水合氯化亚锡。
其中,所述含sn2+的溶液中,所述溶剂可为无水乙醇和/或去离子水。
其中,所述含sn2+的溶液中,所述含sn2+的溶质的浓度可为0.01~10mol/l,例如0.25mol/l或1mol/l。
其中,所述溶解可采用磁力搅拌溶解。
步骤(1)中,优选地,所述聚4-乙烯基吡啶溶液采用下述方法制得:将4-乙烯基吡啶单体或者聚4-乙烯基吡啶和“乙醇和/或水”混合溶解,即得所述聚4-乙烯基吡啶溶液。
其中,所述聚4-乙烯基吡啶溶液中,所述聚4-乙烯基吡啶的单体的浓度优选为0.01~10mol/l,例如1mol/l。
步骤(1)中,所述聚4-乙烯基吡啶的重均分子量可为60000。
步骤(1)中,所述混合的方式可为将所述含sn2+的溶液加入所述聚4-乙烯基吡啶溶液中,也可为将所述聚4-乙烯基吡啶溶液加入所述含sn2+的溶液中。
其中,所述加入的方式可为匀速滴加。
步骤(1)中,所述搅拌形成配合物沉淀的过程中,所述搅拌的时间可为0~48h,例如12h。
步骤(1)中,所述含sn2+的溶液中的sn2+和所述聚4-乙烯基吡啶中的的单体的摩尔比优选为1:20~20:1,例如1:1~1:4,再例如1:1或1:4。
步骤(1)中,所述除去溶剂的方法可为本领域常规的方法,例如蒸发干燥。
步骤(1)中,所述乙醇可为无水乙醇。
步骤(1)中,所述水可为去离子水。
步骤(2)中,所述碳化可采用本领域常规的方法进行,例如将所述碳化前驱体置于石英舟中放入管式炉中,在惰性气氛下进行碳化,即可。
步骤(2)中,所述碳化的升温速率可为1~10℃/min,例如2℃/min。
步骤(2)中,所述碳化的温度可为200~800℃,例如300~400℃,再例如300℃或400℃。
步骤(2)中,所述碳化的保温时间可为0~12h,例如8h。
步骤(2)中,所述碳化的升温过程可为1~10℃/min的升温速率升温至200~800℃,保温0~12h;例如以2℃/min的升温速率升温至300℃,保温8h;再例如以2℃/min的升温速率升温至400℃,保温8h。
步骤(2)中,所述碳化后一般还应进行自然冷却。
本发明还提供了一种氮掺杂碳纳米sno2复合材料,其采用上述方法制得。
本发明还提供了一种氮掺杂碳纳米sno2复合材料,其中,所述氮掺杂碳纳米sno2复合材料包括纳米sno2颗粒和氮掺杂无定形碳骨架;所述纳米sno2颗粒的粒径为3~6nm,所述纳米sno2颗粒中嵌入有复合氧空位,所述复合氧空位的强度为2×106~3×106。
其中,所述复合氧空位的强度可为2×106~2.5×106。
其中,所述复合氧空位的强度可为如图4中sno2@nc所示的复合氧空位强度。
本发明还提供了一种所述氮掺杂碳纳米sno2复合材料作为电池负极材料的应用。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
(1)本发明中以4-乙烯基吡啶与sn2+(例如氯化亚锡)为原料通过锡-氮配位反应生成碳化前驱体,通过原位生长的方式将sno2纳米颗粒直接嵌入到氮掺杂碳的骨架中,有效提高了sno2纳米颗粒的分散性和稳定性。
(2)本发明的复合材料中的sno2纳米颗粒尺寸均一且大小可调,具有制备方法简便、重复性好、可连续生产的特点。
(3)本发明中的氮掺杂碳纳米sno2复合材料电化学性能优异,在能量存储转换、传感、催化等领域有着广泛的应用前景。
附图说明
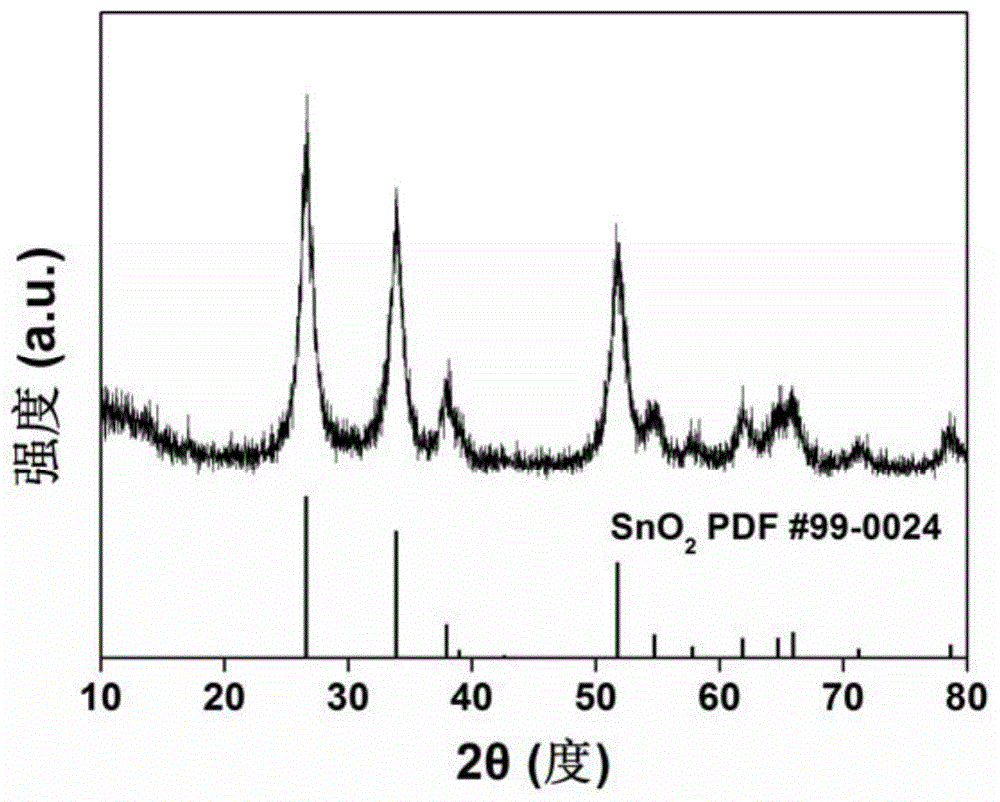
图1为实施例1制备的氮掺杂碳纳米sno2复合材料的x射线衍射图谱(xrd)。
图2为实施例1、2,对比例1、2制备的氮掺杂碳纳米sno2复合材料的透射电子显微镜图像(tem);(a)实施例1,(b)实施例2,(c)对比例1,(d)对比例2。
图3为实施例1制备的氮掺杂碳纳米sno2复合材料的粉末x射线光电子能谱(xps)。
图4为实施例1和对比例1制备的氮掺杂碳纳米sno2复合材料的电子顺磁共振波谱(epr)。
图5为实施例1(sno2@nc)和对比例1(sno2@ncpy)制备的氮掺杂碳纳米sno2复合材料的电子迁移率对比。
图6为实施例1和对比例1制备的氮掺杂碳纳米sno2复合材料的锂离子半电池倍率性能对比。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
下述实施例、对比例所制备的氮掺杂碳纳米sno2复合材料的储锂性能均通过以下方式进行:复合材料的电化学性能测试是以金属锂片作为负极材料在型号为cr2016的纽扣电池中进行的。工作电极的制备过程如下:活性物质、科琴黑和聚偏氟乙烯(pvdf)分别以质量比8:1:1在研钵中以n-甲基吡咯烷酮为分散液进行研磨混合,然后将混合物调成合适粘度的浆料并涂在集流体cu箔上,最后将涂好的cu箔置于真空干燥箱中120℃真空干燥12h。电池测试中的电解液是以碳酸乙烯酯(ec)、碳酸二乙酯(dec)和氟代碳酸乙烯酯(fec)(ec/edc+5%fec)为溶剂的1mlipf6溶液,隔膜型号为celgard2400。整个电池的组装是在ar手套箱中操作。电池测试系统的型号为landct2001a。
下述实施例、对比例中:
聚4-乙烯基吡啶购自sigma-aldrich,4-乙烯基吡啶单体购自百灵威科技有限公司,吡咯单体、无水乙醇和sncl2·2h2o购自greagent。
实施例1
(1)配制聚4-乙烯基吡啶/无水乙醇溶液:室温下,取5.25g聚4-乙烯基吡啶(重均分子量60000)加入到50ml无水乙醇中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的聚(4-乙烯基吡啶)/无水乙醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取11.28gsncl2·2h2o加入到50ml无水乙醇中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl2·2h2o/无水乙醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙醇溶液加到聚4-乙烯基吡啶/无水乙醇溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
实施例2
(1)配制聚4-乙烯基吡啶/无水乙醇溶液:室温下,取5.25g聚4-乙烯基吡啶(重均分子量60000)加入到50ml无水乙醇中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的聚(4-乙烯基吡啶)/无水乙醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取2.82gsncl2·2h2o加入到50ml无水乙醇中,磁力搅拌使其溶解,得到浓度为0.25mol/l的sncl2·2h2o/无水乙醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙醇溶液加到聚4-乙烯基吡啶/无水乙醇溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:4);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
实施例3
(1)配制4-乙烯基吡啶单体/无水乙醇溶液:室温下,取5.25g4-乙烯基吡啶单体加入到50ml无水乙醇中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的4-乙烯基吡啶单体/无水乙醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取11.28gsncl2·2h2o加入到50ml无水乙醇中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl2·2h2o/无水乙醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙醇溶液加到4-乙烯基吡啶单体/无水乙醇溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
实施例4
(1)配制聚4-乙烯基吡啶/无水乙醇溶液:室温下,取5.25g聚4-乙烯基吡啶(重均分子量60000)加入到50ml无水乙醇中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的聚(4-乙烯基吡啶)/无水乙醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取11.28gsncl2·2h2o加入到50ml无水乙醇中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl2·2h2o/无水乙醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙醇溶液加到聚4-乙烯基吡啶/无水乙醇溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至400℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
实施例5
(1)配制4-乙烯基吡啶单体/去离子水溶液:室温下,取5.25g4-乙烯基吡啶单体加入到50ml去离子水中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的4-乙烯基吡啶单体/去离子水溶液。
(2)配制sncl4·5h2o/无水乙醇溶液:取17.53gsncl4·5h2o加入到50ml去离子水中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl4·5h2o/去离子水溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将snso4/去离子水溶液加到4-乙烯基吡啶单体/去离子水溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
对比例1
(1)配制吡咯单体/无水乙醇溶液:室温下,取3.20g吡咯单体加入到50ml无水乙醇中,磁力搅拌使其溶解,得到吡咯单体浓度为1mol/l的聚吡咯单体/无水乙醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取11.28gsncl2·2h2o加入到50ml无水乙醇中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl2·2h2o/无水乙醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙醇溶液加到吡咯单体/无水乙醇溶液中,摩尔比(sn2+:吡咯单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
对比例2
(1)配制聚4-乙烯基吡啶/无水乙二醇溶液:室温下,取5.25g聚4-乙烯基吡啶(重均分子量60000)加入到50ml无水乙二醇中,磁力搅拌使其溶解,得到4-乙烯基吡啶单体浓度为1mol/l的聚(4-乙烯基吡啶)/无水乙二醇溶液。
(2)配制sncl2·2h2o/无水乙醇溶液:取11.28gsncl2·2h2o加入到50ml无水乙二醇中,磁力搅拌使其溶解,得到浓度为1mol/l的sncl2·2h2o/无水乙二醇溶液。
(3)碳化前驱体的制备:室温磁力搅拌条件下,将sncl2·2h2o/无水乙二醇溶液加到聚4-乙烯基吡啶/无水乙二醇溶液中,摩尔比(sn2+:4-乙烯基吡啶单体=1:1);继续搅拌12h,形成稳定的配合物沉淀,再经蒸发干燥即得碳化前驱体。
(4)碳化:将碳化前驱体放置于石英舟内,然后将石英舟放入管式电阻炉,惰性气氛下以2℃/min的升温速率升温至300℃,保温8h然后自然冷却,即可得到氮掺杂碳纳米sno2复合材料。
效果实施例1
按照上述实施例制备氮掺杂碳纳米sno2复合材料,下述效果数据表明通过对本发明中实验条件的调整可以对复合材料的组织结构进行有效调控。
图1是实施例1中氮掺杂碳纳米sno2复合材料的粉末x射线衍射图谱(xrd)。图1说明复合材料中的锡元素物相为sno2。
图2是实施例1、2和对比例1、2中氮掺杂碳纳米sno2复合材料的透射电子显微镜图像(tem),其中,图2a对应实施例1,图2b对应实施例2,图2c对应对比例1,图2d对应对比例2。图2a、图2b说明可通过调整sn2+和4-乙烯基吡啶单体的配比有效调控复合材料中纳米sno2的尺寸,随着sn2+和4-乙烯基吡啶单体的摩尔比的增加纳米sno2的尺寸从3nm(实施例2)增加到6nm(实施例1)。并且,该原位生长的纳米sno2颗粒具有均一的尺寸且均匀的分散在氮掺杂碳基质中。图2c、图2d显示在相同条件下以吡咯单体为配体或以乙二醇为溶剂合成的复合材料中纳米sno2颗粒均具有更大的尺寸,说明配体和溶剂种类对复合材料中纳米sno2尺寸的显著影响。
图3是实施例1制备的氮掺杂碳纳米sno2复合材料的x射线光电子能谱(xps)。图3说明核心元素c,n,sn,o在复合材料表面的含量分别为33.7%,3.5%,12.1%,44.2%。
图4为实施例1和对比例1制备的氮掺杂碳纳米sno2复合材料的电子顺磁共振波谱(epr)。图4说明相比于以吡咯配体,以本发明中的聚4乙烯基吡啶为配体制备的氮掺杂碳纳米sno2复合材料具有更高的氧空位浓度。
图5为实施例1和对比例1制备的氮掺杂碳纳米sno2复合材料的电子迁移率对比。图5表明以本发明中的聚4-乙烯基吡啶为配体制备的氮掺杂碳纳米sno2复合材料sno2@nc的电子迁移率达到357msm-1,远优于以吡咯配体所制备sno2@ncpy的1.01msm-1,也优于背景技术中所述的sno2-x/c复合材料的225msm-1,说明参照本发明实施例制备的复合材料具有更好的导电性,有利于避免由于sno2导电性差所引起的储锂局部反应。
图6为实施例1和对比例1制备的氮掺杂碳纳米sno2复合材料的锂离子半电池倍率性能对比。图6说明作为锂离子电池负极材料,相比于以吡咯配体,以本发明中的聚4乙烯基吡啶为配体制备的氮掺杂碳纳米sno2复合材料表现出更好的倍率性能。在0.1ag-1的电流密度下sno2@nc的可逆容量达到1482mahg-1,即使在10ag-1的高电流密度下sno2@nc仍具有523mahg-1的可逆容量,远高于以吡咯单体为配体制备sno2@ncpy样品的919mahg-1和247mahg-1。
表1
表1为实施例1、2、3、4、5和对比例1、2制备的氮掺杂碳纳米sno2复合材料的储锂性能总结。表1说明实验条件如配体种类(实施例1和对比例1)、溶剂种类(实施例1和对比例2)、原料配比(实施例1和实施例2)、碳化温度(实施例1和实施例4)均对氮掺杂碳纳米sno2复合材料的储锂性能具有显著影响。此外,参考本发明实施样例制备的氮掺杂碳纳米sno2复合材料的储锂容量和初始库仑效率均优于以吡咯单体为配体所制备的氮掺杂碳纳米sno2复合材料。
以上描述了本发明的基本原理、主要特征和优点,而且本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有变化和改进,这些变化和改进都落入要求保护本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!